How DIA-MS Solves Batch Effects and Missing Values in Large-Scale Proteomics
Scaling up proteomic studies from exploratory sets to hundreds of biological samples introduces an often-overlooked challenge: data consistency. In large-cohort research, especially in plasma, serum, or tissue profiling, traditional acquisition strategies frequently result in excessive missing values and unstable quantification across runs. For research teams focused on biological mechanisms or early-phase drug studies, these technical limitations can delay validation, obscure functional pathways, and compromise statistical power.
Data-Independent Acquisition Mass Spectrometry (DIA-MS) offers a fundamentally different acquisition model—one designed to address these systemic bottlenecks in a reproducible, scalable manner.
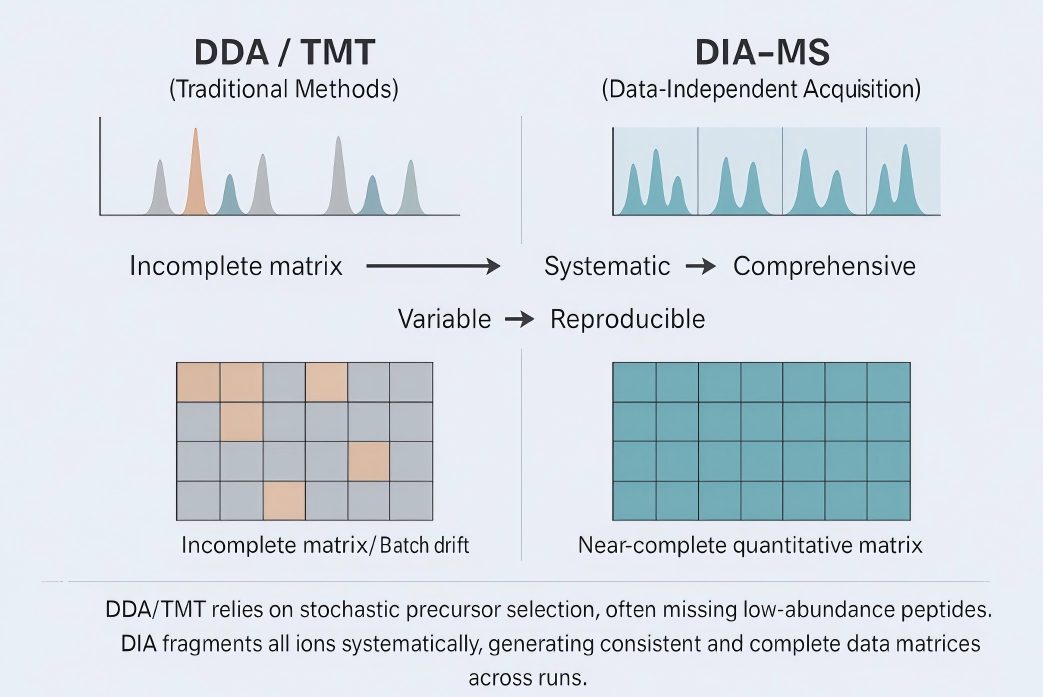 Schematic comparison of acquisition modes. DDA/TMT uses stochastic precursor selection, yielding fragmented matrices; DIA-MS applies fixed windows with all-ion fragmentation, producing near-complete, reproducible data.
Schematic comparison of acquisition modes. DDA/TMT uses stochastic precursor selection, yielding fragmented matrices; DIA-MS applies fixed windows with all-ion fragmentation, producing near-complete, reproducible data.
Why Traditional Acquisition Strategies Fall Short in Cohort-Scale Proteomics
Established methods like Data-Dependent Acquisition (DDA) and tandem mass tag (TMT) labeling remain common in proteomic discovery. However, their performance often declines when applied to large-scale, multi-batch studies. The limitations are structural — not operational.
DDA prioritizes abundant ions in real time, leading to stochastic precursor selection. In small sample sets, this may be tolerable. But across hundreds of samples, the result is inconsistent peptide detection and a fragmented data matrix.
TMT enables relative quantification across multiplexed samples, but its utility weakens beyond a single batch. Bridging multiple TMT sets introduces additional complexity and often results in ratio compression, channel interference, or batch drift.
In both workflows, missing values accumulate, especially for lower-abundance peptides. This compromises statistical modeling, increases false discovery rates, and ultimately obscures biological interpretation — especially in studies aiming to detect subtle mechanistic differences.
Learn more about our Label-Free Quantitative Proteomics Service to see how these limitations compare with data-independent strategies.
What Makes DIA-MS a Structurally Better Solution
Data-Independent Acquisition represents a fundamental shift from selective ion sampling to systematic proteome coverage. In a DIA run, the mass spectrometer fragments all ions within fixed, pre-defined m/z windows, scanning across the entire chromatographic profile. Every peptide signal — not just the most abundant — is consistently captured in every sample.
This comprehensive acquisition model eliminates the randomness of DDA and the batch limitations of TMT. Each sample is subjected to the same acquisition architecture, dramatically improving inter-run consistency. In practice, this means:
- Uniform signal detection across all samples, regardless of batch size or run order;
- Data completeness that exceeds 95% peptide coverage across datasets;
- Low coefficient of variation (CV) and reduced dependency on missing-value imputation.
For researchers working on mechanistic insights or early-stage target validation, this translates to greater statistical power and confidence in observed trends — not because more data is generated, but because fewer signals are lost.
Our DIA Quantitative Proteomics Service leverages SWATH-type acquisition and curated spectral libraries to deliver high-throughput proteomic data with built-in consistency.
The Statistical Advantage of Complete Proteomic Matrices
In quantitative proteomics, data completeness is not just a convenience — it is a prerequisite for valid interpretation. When missing values exceed manageable thresholds, core statistical tools like PCA, clustering, and differential expression lose power and become biased. This is especially critical in preclinical models where fold changes may be modest and biological variability low.
DIA-MS addresses this problem directly. By capturing all detectable peptides across every sample, it generates highly complete data matrices — often with missing value rates below 5%. This level of data integrity enables:
- Reliable statistical modeling using LIMMA, ANOVA, or mixed linear models without relying on heavy imputation;
- Improved sensitivity in detecting biologically relevant changes, especially among low-abundance targets;
- Robust multi-omics integration, as complete proteomic datasets align more accurately with transcriptomic and metabolomic layers.
For research teams working on time-course studies, compound mechanism profiling, or signaling network reconstruction, this level of statistical completeness unlocks a new layer of interpretability.
How DIA Is Transforming Large-Cohort Biomarker Discovery
While DIA-MS is gaining recognition in biomarker discovery, its greatest impact may lie upstream — in the basic and pre-clinical research stages, where reproducibility and biological nuance are paramount.
Fast runs without shallow data.
If you need to move hundreds of plasma or cell samples and still read real biology, the Scanning SWATH variant of DIA is a practical lever. It compresses gradients and speeds duty cycles yet still tracks mechanism and disease signals. One Nature Biotechnology study used Scanning SWATH to profile drug modes of action and plasma proteomes; notably, it validated 43 and discovered 11 additional plasma protein markers associated with COVID-19 severity, showing that high-throughput does not have to trade off with signal fidelity.
Why this matters: time-course and multi-arm designs become feasible on normal lab schedules—while preserving the peptide coverage you need for classifiers and longitudinal models.
Making multi-batch and multi-site data play nicely.
Large programs rarely finish in a single analytical block. Here, DIA's dense, uniform matrices enable effective harmonization rather than rescue by imputation. The STAVER framework demonstrated this at scale: by standardizing against reference datasets, it recovered consistent plasma biomarker signals across four independent DIA COVID-19 cohorts and a colorectal cancer cohort, despite platform and site differences. In other words, the method reduces unwanted variation so biology shows through.
Why this matters: replication across labs—and merging historical with new batches—becomes statistically defensible, not a source of drifting cut-offs.
Deep plasma with realistic budgets.
A frequent worry is that going deep in plasma means sacrificing scale. A recent Journal of Proteome Research study outlined a streamlined workflow that pairs moderate prefractionation with DIA to substantially increase coverage while staying cost-conscious—explicitly designed for discovery cohorts rather than boutique datasets. A companion preprint details similar gains and discusses biomarker identification on top of these deeper matrices.
Why this matters: you can profile the tricky, low-abundance layers—immune, ECM, signalling—without blowing out run time or consumables.
From protocol to practice: fewer missing values, steadier stats.
Teams planning large clinical or pre-clinical cohorts now have a protocolised playbook. A Nature Protocols guide on SWATH/DIA emphasises that, at cohort scale, DIA yields markedly more reproducible protein measurements than DDA and "greatly alleviates the missing-data problem." It also codifies QC layout, window schemes, and normalisation—exactly the operational detail project teams ask for.
Why this matters: cleaner inputs for LIMMA/ANOVA and machine learning; fewer artifacts from imputation; higher cross-batch R².
Libraries that broaden the candidate space.
Discovery benefits from a wide, stable search space. The DPHL v2 pan-human DIA library expands coverage across 24 sample types, increasing identified and differentially regulated proteins compared with v1—practical evidence that better libraries widen the pool of candidate biomarkers and improve identification stability across cohorts.
Why this matters: better coverage today means fewer "missing targets" tomorrow, even as batches and sites change.
How DIA-MS Delivers Superior Data Integrity
Large-scale proteomics depends not just on depth, but on trustworthy data structures that remain stable across hundreds of injections. DIA-MS achieves this by making consistency an intrinsic property of acquisition rather than an afterthought of correction. Instead of adjusting for missingness later, the method prevents it at the source.
Across studies, DIA routinely achieves >95 % peptide coverage, <5 % missing values, and cross-batch R² above 0.95. This consistency ensures that what you compare across time points or treatment arms reflects real biology, not analytical drift. Statistical tools—from clustering and PCA to LIMMA and mixed-effect models—operate with full data matrices, yielding stronger, more reproducible insights.
For complex sample types such as plasma or tissue lysates, DIA's uniform acquisition and curated spectral libraries also improve signal-to-noise stability, variance control, and multi-omics integration. Researchers gain clearer trends, smaller error bars, and validated mechanisms that hold up under replication.
| Metric | DDA (Traditional) | DIA (Data-Independent) |
| Quantifiable Proteins | ~2,000 | >4,000 |
| Missing Value Rate | 35–40% | <5% |
| Cross-Batch Correlation (R²) | 0.70–0.80 | >0.95 |
| Coefficient of Variation (CV) | 20–25% | <10% |
| Data Matrix Completeness | Partial | Near-Complete |
These gains are not incremental. They redefine what's achievable in large-cohort proteomics, especially for studies involving low-abundance targets, complex matrices, or long analytical timelines. For most projects exceeding 300 samples, DIA is no longer a technical preference — it's a statistical necessity.
References
- Messner, Christoph B., et al. "Ultra‑fast proteomics with Scanning SWATH." Nature Biotechnology 39.7 (2021): 846‑854. https://doi.org/10.1038/s41587-021-00860-4
- Ran, Peng, et al. "STAVER: a standardized benchmark dataset-based algorithm for effective variation reduction in large-scale DIA-MS data." Briefings in Bioinformatics 25.6 (2024): bbae553. https://doi.org/10.1093/bib/bbae553
- Ward, Bradley, et al. "Deep plasma proteomics with data-independent acquisition: Clinical study protocol optimization with a COVID-19 cohort." Journal of proteome research 23.9 (2024): 3806-3822. https://doi.org/10.1021/acs.jproteome.4c00104
- Shen, Shichen, et al. "High-quality and robust protein quantification in large clinical/pharmaceutical cohorts with IonStar proteomics investigation." Nature protocols 18.3 (2023): 700-731. https://doi.org/10.1038/s41596-022-00780-w
- Xue, Zhangzhi, et al. "DPHL v. 2: An updated and comprehensive DIA pan-human assay library for quantifying more than 14,000 proteins." Patterns 4.7 (2023). https://doi.org/10.1016/j.patter.2023.100792
- Zhang, S., Chen, W., Lin, Y., et al. "Deep Plasma Proteomics with Data‑Independent Acquisition: Clinical Summary and Biomarker Discovery." Journal of Proteome Research (2024). https://doi.org/10.1021/acs.jproteome.4c00104
- Yu, Hancheng, Zhang, Jijuan, Qian, Frank, et al. "Large‑Scale Plasma Proteomics Improves Prediction of Peripheral Artery Disease in Individuals With Type 2 Diabetes: A Prospective Cohort Study." Diabetes Care 48.3 (2025): 381‑389. https://doi.org/10.2337/dc24‑1696




 4D Proteomics with Data-Independent Acquisition (DIA)
4D Proteomics with Data-Independent Acquisition (DIA)