DIA Proteomics for Biobanks and Population Cohorts: Building Reusable Data Assets for the Next Decade
A plasma or serum collection that starts as a cardiovascular project may later support oncology, neurology, infectious disease, or aging research. New outcomes appear as follow-up continues, new omics layers are added, and new analysis methods arrive.
If you invest in proteomics on such a resource, you don't just want "a dataset for this year." You want a proteomics layer that can be reused, extended, and re-interpreted over the next decade.
Data-independent acquisition (DIA) mass spectrometry is increasingly chosen for large cohorts because it delivers deep coverage, low missingness, and strong reproducibility across many plasma or serum samples—while preserving raw data that can be re-analyzed with new libraries and algorithms later.
This resource explains how to use DIA proteomics for biobanks and population studies, and how the NGPro™ platform helps teams turn one-time MS runs into long-lived, cohort-grade data assets.
Why Biobanks and Population Cohorts Need Archivable Proteomics Data
From one-off experiments to long-lived data assets
Traditional proteomics projects are often scoped like this:
- One disease endpoint
- One fixed set of samples
- One analysis pipeline, used once and then archived
Biobanks and long-running cohorts work very differently:
- Follow-up accumulates over years – new events (e.g., stroke, dementia, cancer) appear over time.
- New phenotypes and subtypes are defined – multimorbidity, polypharmacy, frailty, molecular subtyping.
- Analytical tools evolve – new DIA algorithms, improved spectral libraries, and better integration methods emerge.
This means the proteomics layer should behave like a persistent data asset, not a frozen snapshot. It should support questions such as:
- Can we revisit the same raw data when new outcomes are adjudicated?
- Can we re-process the files with improved libraries or software?
- Can we add new sample waves later, without starting from zero?
Practical constraints unique to large cohorts
Biobank and cohort teams face constraints that are different from a typical lab-scale experiment:
- Tens of thousands of stored aliquots, but limited LC–MS capacity
- Multiple collection centers with different staff and workflows
- Staged funding – pilot now, expansion later
- Strong pressure to align with genomics, metabolomics, imaging, and clinical data models
A suitable proteomics approach must therefore:
- Work well in waves (nested case–control, case–cohort, sub-cohorts)
- Tolerate multi-year, multi-center generation of data
- Produce clean protein × sample matrices that downstream statisticians and epidemiologists can use without rewriting their toolkits
Limitations of Classical DDA and TMT/iTRAQ in Long-Term Cohort Projects
DDA: strong for small discovery sets, fragile for staged cohorts
In data-dependent acquisition (DDA), the instrument continuously selects the most intense precursor ions for fragmentation in each cycle. This is excellent for exploratory, small-scale discovery—but for large or staged cohort studies it introduces issues:
- Stochastic precursor selection – low-abundance peptides may be sampled in some runs but missed in others.
- High and structured missingness – especially problematic when combining multiple batches or waves.
- Limited flexibility for extension – adding a second wave of samples years later often leads to matrices with different missing-value patterns and depth.
In practice, DDA-based cohort projects can run into:
- Difficulties merging "old" and "new" sub-cohorts
- Heavier reliance on imputation and aggressive filtering
- Reduced confidence when deriving risk scores or mechanistic signatures from incomplete matrices
TMT/iTRAQ: powerful multiplexing, rigid long-term design
Isobaric labeling (TMT/iTRAQ) can multiplex many samples within a single run, which is attractive for tightly scoped studies. In population cohorts, however, other issues dominate:
- Rigid batch structures – once plex layouts and bridge channels are set, later additions are hard to integrate seamlessly.
- Ratio compression in complex matrices, especially plasma/serum.
- Operational complexity – multi-plex, multi-batch designs need careful balancing and are vulnerable to label or instrument issues.
Retrospective re-use becomes difficult: extending the project usually means building new plexes with their own batch effects, not simply appending rows to an existing matrix.
What cohort leaders actually ask for
From discussions with cohort and biobank teams, requirements usually sound like this:
- "We want to profile a subset now, but keep options open for future outcomes."
- "We need raw data we can re-process with new tools—not just one frozen analysis."
- "We want a statistically friendly matrix that we can join to our genomics and clinical data without heroics."
These priorities align closely with what DIA proteomics can provide.
DIA Proteomics as a Foundation for Reusable Cohort Data
Systematic coverage and less missingness
In DIA, the m/z range is divided into predefined windows, and all ions in each window are fragmented on every cycle. Instead of chasing only the top N precursors, DIA builds a more complete fragment-ion record for the entire chromatographic run.
For biobanks and cohorts, this translates into:
- More systematic sampling across runs and batches
- Lower missing value rates at the protein level, even in plasma and serum
- Improved run-to-run reproducibility, which is crucial when projects span months or years
The result is a data structure that behaves much more like a broad, quantitative feature matrix, making it easier to:
- Run regression, survival, and mixed models
- Build protein-based risk scores
- Perform clustering, network analysis, and pathway enrichment across large groups
For a deeper technical comparison between DIA and DDA, see our resource DIA vs DDA Mass Spectrometry: A Comprehensive Comparison.
Library-based and library-free DIA for long-term reuse
DIA data can be interpreted using empirical spectral libraries (generated by DDA or GPF-DIA) or by library-free (direct) DIA workflows that rely on in silico prediction and advanced scoring.
For biobanks, this matters because:
- High-quality libraries built from representative cohort samples can be reused whenever new endpoints or sub-cohorts are analyzed.
- Library-free tools make it possible to apply new algorithms to old raw files, improving identifications and quantification without repeating LC–MS.
If you are planning a long-running cohort program, the article Library-Free vs Library-Based DIA Proteomics: Strategies, Software, and Best Use Cases is a useful companion resource.
Re-analysis as knowledge and tools evolve
Because DIA captures fragment-ion information across the full acquisition window, re-analysis is built into the data structure. In practice, that means:
- When a new clinical endpoint is defined (e.g., a specific cancer subtype or neurodegenerative disease), the same raw DIA files can be processed again, now using the new case/control labels.
- When improved software (e.g., updated DIA-NN, Spectronaut, FragPipe workflows) becomes available, you can re-run pipelines and extract more or better-scored peptides from the same data.
- When new pathway databases or protein panels become relevant, you can generate additional targeted outputs from existing DIA matrices.
This is exactly the behavior cohort teams expect from a "run once, analyze for years" proteomics strategy.
Study Design Considerations for Biobank and Epidemiology DIA Projects
Sampling strategies: nested designs and sub-cohorts
In large cohorts, you rarely send every stored sample to the mass spectrometer. Instead, teams use designs familiar from epidemiology:
- Nested case–control – incident cases plus matched controls from within the cohort
- Case–cohort – all cases plus a randomly selected sub-cohort
- Disease-specific sub-cohorts – for example, all participants with type 2 diabetes plus a reference group
DIA fits naturally into these strategies:
- You can begin with a well-powered discovery subset (hundreds of subjects) to establish feasibility and signatures.
- As new outcomes or questions appear, you can add additional nested sets, keeping acquisition and analysis consistent.
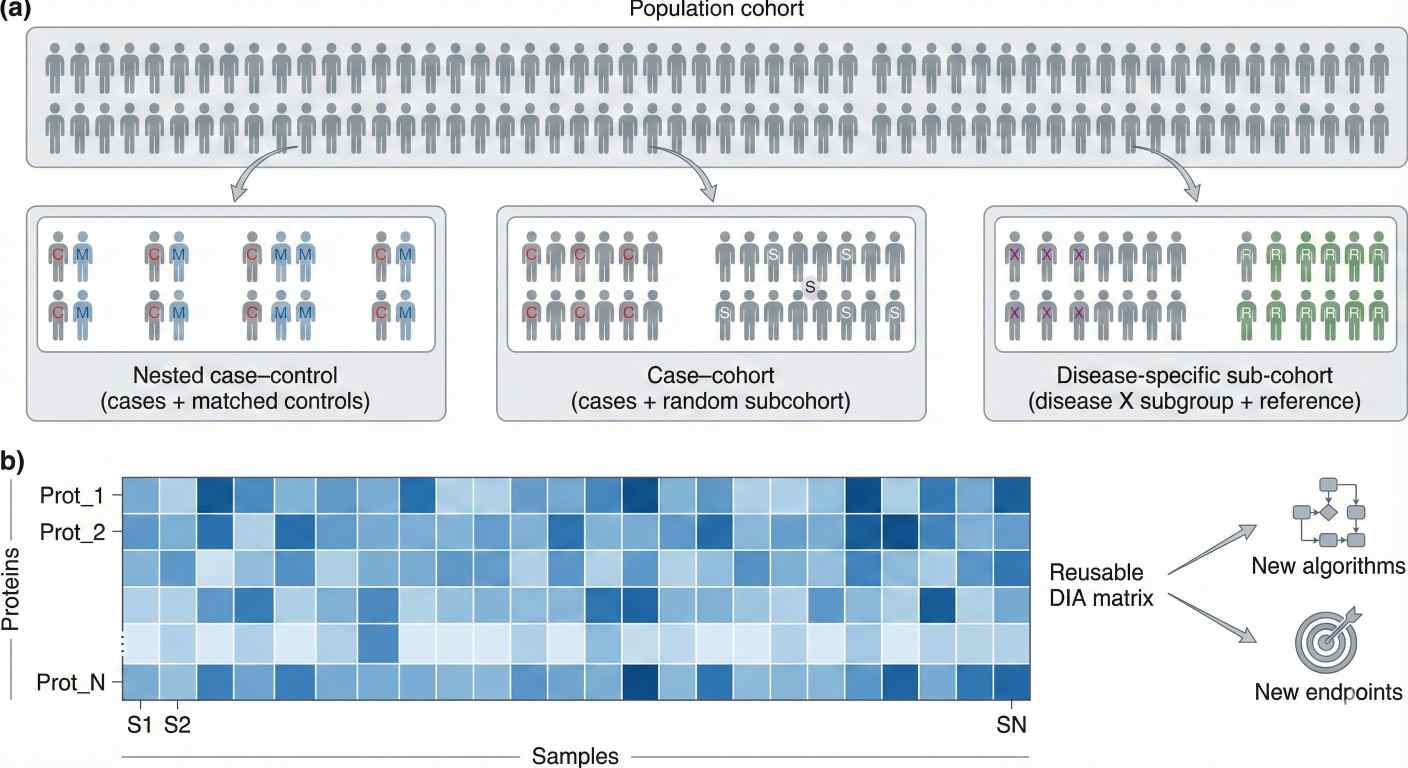 Sampling strategies in a population cohort and the resulting DIA proteomics protein × sample matrix for reuse across multiple analyses.
Sampling strategies in a population cohort and the resulting DIA proteomics protein × sample matrix for reuse across multiple analyses.
Batch and platform planning for long-term extension
For biobank projects, it is worth planning upfront how new waves of samples will connect to baseline data. DIA does not remove the need for good experimental design—but it does make extensions more manageable.
A simplified planning view:
| Design Question | DIA-Focused Consideration for Cohorts |
| How many samples in the first wave? | Start with a powered subset; design methods that can be reused later. |
| How often will new waves be added? | Keep gradients, windows, and QC structure compatible over time. |
| Will instruments or labs change? | Use shared reference/QC samples and overlapping designs to bridge. |
| How will pre-analytic factors be tracked? | Capture freeze–thaw cycles, storage time, and site in metadata. |
Label-free DIA is especially attractive here because it decouples sample numbers from fixed labeling plexes. The Label-Free DIA Quantitative Proteomics service is built around this flexible, scalable model.
For cohorts that require deeper coverage or shorter LC gradients—such as very large plasma panels—the 4D-DIA Quantitative Proteomics Services use ion mobility separation to boost sensitivity and data completeness, particularly in large clinical cohorts.
Sample types and biobank realities
Biobanks frequently store:
- Plasma or serum (often at multiple visits)
- Urine and other biofluids
- PBMCs or buffy coat
- Occasionally FFPE tissues or specialized fluids
The core DIA principles do not change, but sample preparation quality becomes the main determinant of success. Consistent extraction, digestion, and cleanup across centers is essential to maintain longitudinal and cross-section comparability.
For a deeper, practical guide to preparing large sets of samples for DIA, see Optimizing Sample Preparation for DIA Proteomics: Best Practices.
Building an Archivable Proteomics Layer with the NGPro™ Platform
The NGPro™ platform is designed to support exactly this type of longitudinal, cohort-style work—combining discovery and targeted workflows under a DIA-first strategy.
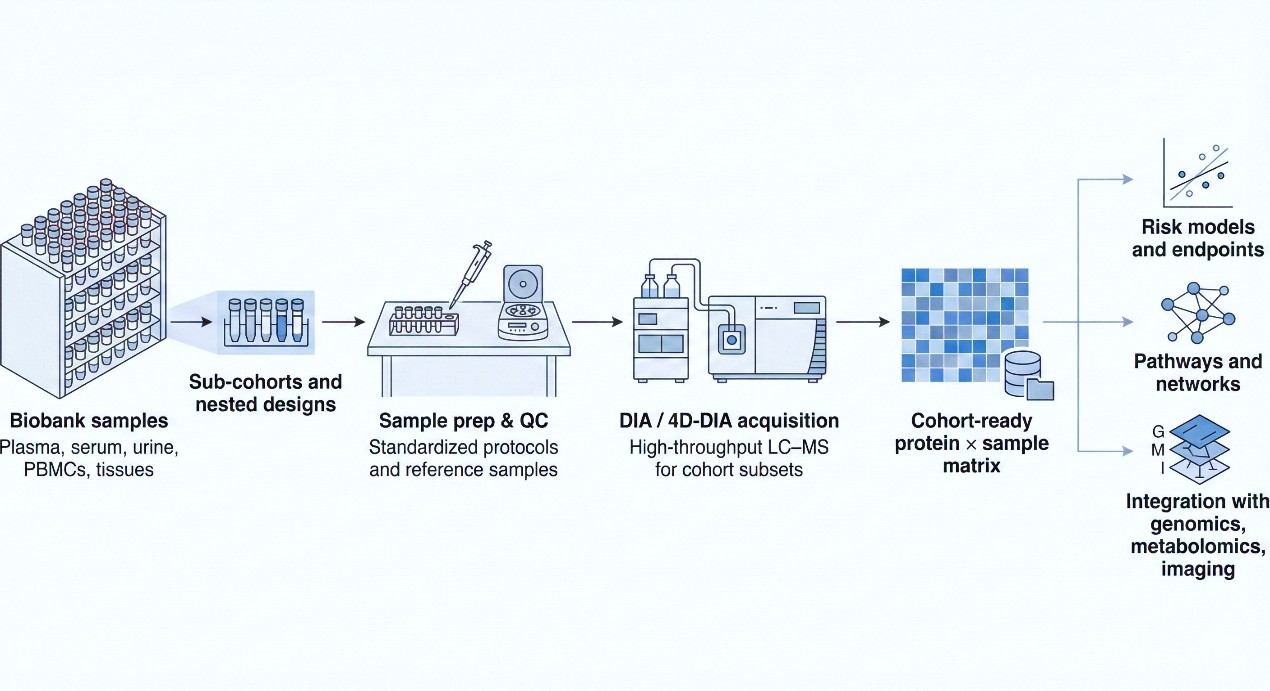 Workflow for applying DIA proteomics to biobank samples, from sub-cohort selection and sample prep through DIA/4D-DIA acquisition to reusable cohort-level data and downstream analyses.
Workflow for applying DIA proteomics to biobank samples, from sub-cohort selection and sample prep through DIA/4D-DIA acquisition to reusable cohort-level data and downstream analyses.
Step 1 – Cohort consultation and long-range planning
Early scoping queries often include:
- Which diseases and outcomes are already in focus, and which are likely to be important in 5–10 years?
- Which sampling strategy (nested case–control, case–cohort, sub-cohort) provides the best balance of power and cost?
- What is the right mix of standard DIA, 4D-DIA, and GPF-DIA for this cohort?
- How will proteomics data link to genomics, metabolomics, and imaging in downstream models?
The goal is to ensure that the proteomics strategy fits the lifetime of the cohort, not just the first manuscript.
Step 2 – Standardized sample handling and QC across centers
In multi-center or multi-year projects, NGPro emphasizes:
- Harmonized protocols for thawing, aliquoting, and extraction
- Matrix-appropriate workflows for plasma, serum, urine, PBMCs, and tissues
- Embedded reference and QC samples to monitor batch and instrument behavior over time
The same DIA-centric principles that underpin the Discovery Proteomics Service are applied, but tailored to cohort scale and epidemiological constraints.
Step 3 – DIA / 4D-DIA acquisition for cohort subsets
Depending on depth and throughput requirements:
- Standard DIA runs can quickly generate robust matrices across hundreds of participants.
- 4D-DIA or GPF-DIA can be reserved for key sub-cohorts or more demanding questions, e.g., deeper coverage in a particular disease group.
Here, the DIA Quantitative Proteomics Service often serve as the main acquisition engines for population-style datasets.
Step 4 – Cohort-level DIA data analysis and delivery
For large DIA cohorts, data analysis is its own project—not a side task. The Data Independent Acquisition (DIA) Data Analysis service combines spectrum-centric and peptide-centric strategies to:
- Build or refine spectral libraries (including from GPF-DIA)
- Control FDR at peptide and protein levels
- Generate consistent protein × sample matrices with clear QC metrics
- Provide outputs that are ready to join with genotypes, metabolomics, and clinical variables
Rather than returning only differential lists, the focus is on delivering structured, reusable data assets that can support multiple analytic waves.
Step 5 – Target panels and follow-up studies
Discovery-scale DIA typically reveals more candidates than can be followed up in every future wave. To translate these findings into practical tools, We offer Targeted Proteomics Services.
Typical next steps:
- Distill broad signatures into short, robust protein panels
- Validate behavior in independent sub-cohorts or partner biobanks
- Deploy targeted assays in extended cohorts or translational pipelines
For more context on how NGPro uses DIA in discovery-scale projects, see Quantitative Discovery Proteomics with DIA: Depth, Consistency, and Scale.
Example Use Patterns in Biobank and Population Studies
Below are example patterns showing how DIA proteomics is actually used in cohort settings.
Cardiovascular nested case–control in a population cohort
Scenario
A cardiovascular cohort with >20,000 participants and plasma stored at multiple visits wants to understand proteomic signatures of incident heart failure and major adverse cardiovascular events.
Approach
- Define nested case–control sets for specific endpoints (e.g., heart failure, myocardial infarction), matching on age, sex, and baseline risk factors.
- Run DIA plasma proteomics on these subsets using a standardized acquisition and QC design.
- Model associations between protein signatures, traditional risk factors, and outcomes, as in recent population-scale plasma proteomics work.
Expert view
- Treat each nested set as part of a coherent DIA program, not stand-alone experiments.
- Pre-define how new endpoints (e.g., atrial fibrillation or kidney disease) will reuse the same acquisition and analysis framework.
Longitudinal multi-disease risk profiling
Scenario
A general population study wants to evaluate how early proteomic patterns relate to later risk across several disease categories—oncology, neurodegeneration, and metabolic disease.
Approach
- Choose a multi-disease panel of endpoints and design an appropriate case–cohort or multi-endpoint nested design.
- Use DIA to measure plasma proteomes at key timepoints (e.g., baseline and mid-follow-up).
- Train protein-based scores that complement established risk scores and examine their performance across diseases.
Expert view
- Plan analytical strategies that allow re-use of the same DIA features across multiple disease models, rather than building totally separate pipelines per endpoint.
- Reserve the option of a second proteomics wave at a later visit, with matched DIA methods, to study temporal change.
Multi-omics integration in population health
Scenario
A cohort provides genome-wide genotype data, metabolomics, and imaging. Proteomics is expected to act as the key "bridge layer" between genotype and phenotype.
Approach
- Apply DIA proteomics on a sub-cohort with full multi-omics coverage.
- Perform pQTL analysis and integrate proteins with metabolites and clinical traits.
- Use multi-omics models to identify mechanisms linking genetic variation, protein networks, and phenotypes.
Expert view
- Decide early on ID namespaces, scaling, and batch handling for all omics layers.
- Treat study design as the first integration step; a detailed guide is available in How to Integrate DIA Proteomics Data with Multi-Omics Platforms.
When DIA Is – and Isn't – the Right Choice for Biobanks
DIA is particularly well-suited when:
- You need broad, unbiased molecular coverage across hundreds or thousands of samples.
- You expect to revisit and extend the dataset as new outcomes or tools emerge.
- You plan multi-omics integration and require a robust proteomics layer that behaves like a quantitative feature matrix.
Other approaches may be preferable when:
- You already have small, fixed protein panels and need ultra-high-throughput targeted assays only.
- The goal is a narrow, one-time study with no expectation of later re-analysis or expansion.
In many biobank programs, a blended strategy works best:
- Use DIA to build an initial discovery-grade proteomics layer on carefully chosen subsets.
- Identify stable markers and signatures.
- Transition to targeted panels (PRM/SRM or immunoassays) for large-scale deployment in extended cohorts or clinical environments.
Frequently Asked Questions (FAQ)
Yes. DIA proteomics has been successfully applied in large clinical cohorts and biobank-style studies, precisely because it scales from hundreds to thousands of samples with standardized methods and low missingness.
In practice, feasibility is driven less by the total size of the biobank and more by how you design waves or sub-cohorts (e.g., nested case–control or case–cohort) and keep acquisition and analysis consistent over time.
DIA raw files contain fragment-ion information for all ions within the acquisition windows, so they can be re-processed with new spectral libraries or updated software without repeating LC–MS runs.
When new clinical endpoints are defined or new algorithms are released, you can rerun the analysis pipelines to generate updated protein matrices and statistics, as long as metadata and QC have been maintained.
Most cohorts use a staged strategy rather than analyzing all participants at once. Effective options include nested case–control, case–cohort, and disease-specific sub-cohorts.
A practical approach is to start with a powered subset aligned to one or two endpoints, evaluate performance, and then extend the same DIA design to additional outcomes or follow-up waves.
Batch control relies on a combination of standardized sample preparation, embedded QC/reference samples, and statistical normalization in a consistent DIA analysis pipeline.
For long-running projects, it is common to monitor instrument performance metrics, analyze pooled QC samples regularly, and apply normalization or batch-correction methods before epidemiological or multi-omics modeling.
Yes. DIA produces quantitative protein matrices that can be linked to other omics and clinical data via shared IDs and harmonized scaling.
Successful integration usually requires:
- Consistent specimen IDs and visit times across platforms,
- Harmonized formats and QC thresholds, and
- Integration methods (feature-level, latent-space, or network-based) suited to cohort size and missingness patterns.
References
- Lou, Ronghui, and Wenqing Shui. "Acquisition and analysis of DIA-based proteomic data: A comprehensive survey in 2023." Molecular & Cellular Proteomics 23.2 (2024): 100712.
- Fröhlich, Klemens, et al. "Data-independent acquisition: A milestone and prospect in clinical mass spectrometry–based proteomics." Molecular & Cellular Proteomics 23.8 (2024): 100800.
- Ward, Bradley, et al. "Deep plasma proteomics with data-independent acquisition: Clinical study protocol optimization with a COVID-19 cohort." Journal of Proteome Research 23.9 (2024): 3806–3822.
- Ou, Guanyong, et al. "Deep data-independent acquisition-based plasma proteomic profiling unveils distinct molecular features in dengue fever with neutropenia." Virologica Sinica (2025): Article in press.
- Lieb, Wolfgang, et al. "Population-based biobanking." Genes 15.1 (2024): 66.

 4D Proteomics with Data-Independent Acquisition (DIA)
4D Proteomics with Data-Independent Acquisition (DIA)