Translational Research Benchmark
Predicting immune-related adverse events (irAEs) via Proteome-Wide Profiling
Scientific Objective & Challenge
Immune checkpoint inhibitors (ICIs) have radically transformed oncology, yet a significant proportion of patients experience severe immune-related adverse events (irAEs) such as myocarditis. The underlying mechanisms remained poorly understood, and traditional targeted immunoassays failed to identify reliable pre-treatment predictors. Researchers sought to uncover the underlying autoantibody profiles by retroactively screening pre-treatment sera from a large patient cohort (200+ samples).
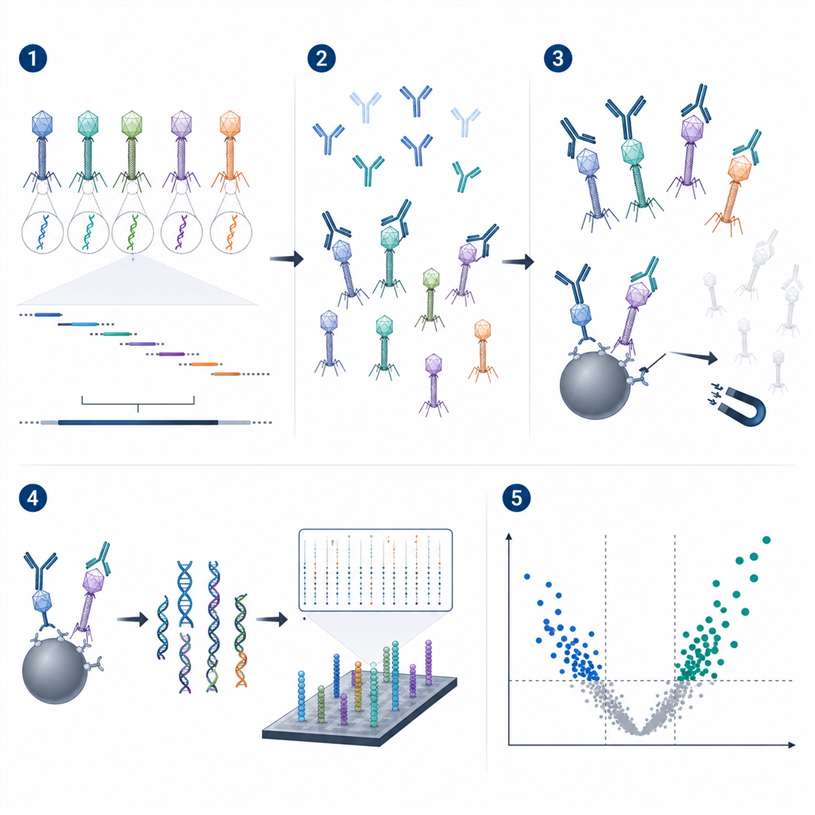
PhIP-Seq Experimental Design
- Utilized a comprehensive 90-mer Human Peptidome library with a 45-amino acid overlap.
- Ensured robust coverage of linear and local secondary epitopes across the entire proteome.
- Implemented automated robotic immunoprecipitation to ensure standardized stringency, followed by deep NGS sequencing.
Transformative Discoveries
- Pre-treatment Detection: Unbiased screening identified trace-level, pre-existing autoantibodies invisible to standard arrays.
- Target Localization: Specific motifs on tissue-restricted proteins correlated with localized toxicities (e.g., myocarditis).
- Resolution: Bioinformatics narrowed binding sites to precise 7-amino acid minimal motifs.
Research Impact
Prediction precision of severe irAEs prior to ICI therapy
The deployment of PhIP-Seq provided the structural resolution necessary to move beyond candidate-gene guesswork. It established a robust biomarker discovery framework, demonstrating that high-throughput profiling of the autoantibody repertoire can uncover critical mechanisms driving immunotoxicities, paving the way for future validation studies in oncology.
Scientific Reference
Shrock, E., et al. (2023). Germline-encoded amino acid–binding motifs drive immunotoxicities. Science. PMC10874550.