Can the bands on SDS gels be cut and sent for mass spectrometry sequencing?expand_more
Yes, bands separated by SDS can be directly cut and sent for mass spectrometry identification. To enhance identification accuracy, it is important to use an appropriate gel concentration for separating the target protein bands and to separate neighboring protein bands from the target protein as much as possible during gel electrophoresis.
Can protein bands on SDS gels that are too thin be identified?expand_more
Generally, for protein bands stained with Coomassie or SYPRO Ruby, bands visible to the naked eye are sufficient for sequencing.
What if the cut SDS protein bands contain more than one band?expand_more
If the cut protein bands contain multiple bands, they can still be analyzed for protein identification. Protein sequences can subsequently be inferred through data comparison. In fact, most protein bands cut from SDS-PAGE gels contain multiple proteins. These complex proteins can be analyzed through chromatography-mass spectrometry tandem identification methods, where complex proteins are first separated by chromatography and then analyzed one by one through mass spectrometry, achieving higher accuracy.
Can Western blot results be used for protein sequencing?expand_more
Proteins transferred after Western blotting can indeed be sequenced. By comparing Western blotting results to SDS-PAGE gels, the corresponding positions of protein bands on SDS-PAGE gels can be identified and then cut for mass spectrometry identification. If the protein quantity is sufficient and the purity is high, protein bands on PVDF membranes can be directly cut for N-terminal sequencing identification.
What precautions should be taken during protein sample preparation?expand_more
Mass spectrometry for protein identification is highly sensitive, and even trace amounts of contaminating proteins introduced during the operation can be detected, significantly affecting the accuracy of protein sequencing analysis. Therefore, during sample preparation, clean and uncontaminated vessels, reagents meeting mass spectrometry purity requirements, freshly prepared solutions, gloves, and head covers should be used to avoid contamination by keratin and other contaminants.
What are the requirements for preparing various types of samples for protein sequencing?expand_more
In general, protein samples separated by SDS-PAGE gels and stained with Coomassie or SYPRO Ruby are compatible with mass spectrometry identification. However, protein samples stained with silver must not use glutaraldehyde as a fixative, as it would affect subsequent mass spectrometry analysis. Additionally, when the protein concentration in solution is low, the use of surfactants like SDS should be minimized, and salt concentration should be reduced to improve identification accuracy.
How to prepare protein samples if the molecular weight of the protein of interest is small?expand_more
If the molecular weight of the protein of interest is relatively small, high-concentration SDS-PAGE gels can be used to separate proteins, followed by staining with Coomassie, and then cutting gel bands consistent in size with the target protein for mass spectrometry identification.
What are the requirements for sample shipment?expand_more
Protein bands and powder are relatively stable and can be transported using ice packs. Protein solution should be shipped using dry ice. It is recommended to lyophilize the samples before shipping, as proteins are highly stable in lyophilized form. Repeated freeze-thaw cycles should be avoided to prevent protein degradation.
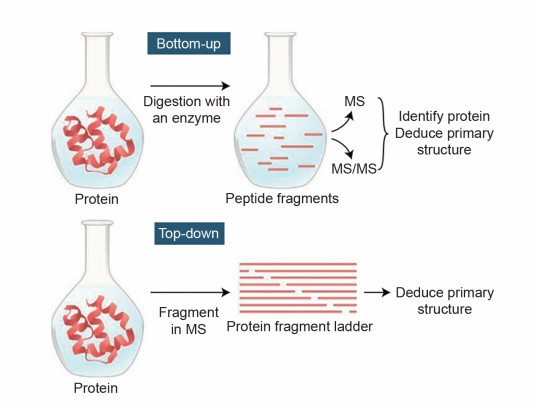
Why is it necessary to enzymatically digest proteins into peptides before mass spectrometry sequencing?expand_more
The larger the protein fragments, the lower the accuracy of mass spectrometry detection. Therefore, before mass spectrometry detection, proteins need to be digested into smaller peptides to improve detection accuracy. Generally, peptides with 6-20 amino acids are most suitable for mass spectrometry detection.
How to improve the efficiency of determining the sequence of modified peptide segments in protein sequencing?expand_more
Under current conditions, it is difficult for mass spectrometry to provide 100% sequence coverage of peptides. Some peptides may be lost during the process, and protein phosphorylation can inhibit trypsin digestion. Moreover, phosphorylated peptides are much less abundant than non-phosphorylated peptides, which may inhibit the mass spectrometry response. Therefore, efforts should be made to minimize non-phosphorylated peptides. Methods such as fractionation, IMAC, and antibody binding can be used. MALDI-TOF-MS can be used to determine the molecular weight of peptide segments; if the measured mass is 80 Da or its multiples higher than expected, phosphorylation can be inferred.
What could cause two amino acid residues to undergo Edman degradation simultaneously in the determination of protein N-terminal amino acid sequences?expand_more
When two amino acid residues are detected simultaneously in Edman degradation, it is possible that the protein is impure and contaminated with other proteins. If one of the amino acids is glycine, residual glycine from the buffer in the protein band may not have been completely removed, leading to this result.