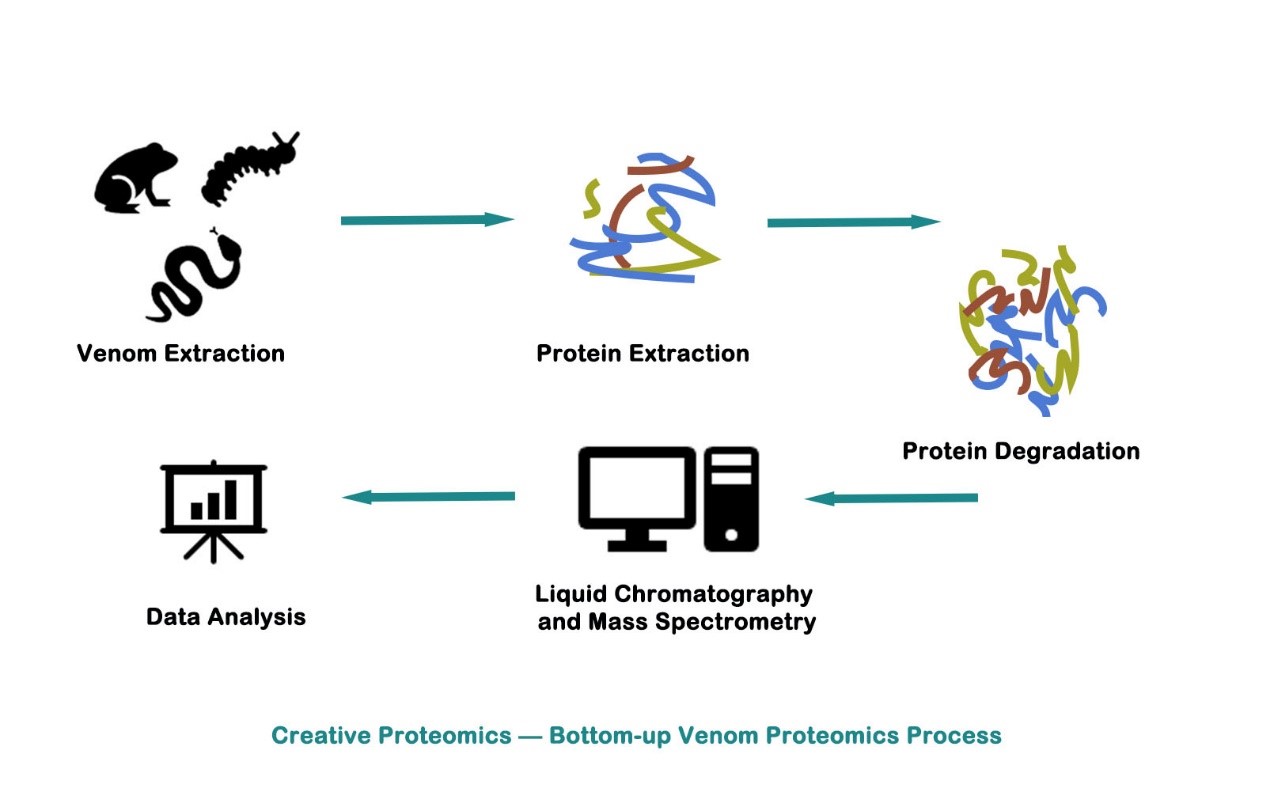
Bottom-up Proteomics in Venom
Animal venom is a potential natural resource with potential applications in medicine, agriculture and industry. Clarifying the toxins in venom is an important step in venomics research. Modern venom research usually uses proteomics because the targets in these mixtures are peptides and/or proteins. Proteomics plays an important role in identifying venom toxins, determining their main structures (including post-translational modifications), and studying the physiological mechanisms of their production and transmission.
Qualitative protein analysis usually refers to the use of mass spectrometry for protein identification and sequence analysis. Qualitative protein analysis is divided into two strategies: top-down analysis and bottom-up analysis. Bottom-up, or Shotgun strategy, is currently the mainstream strategy. Bottom-up proteomics combines high-performance liquid chromatography (HPLC) technology and mass spectrometry (MS). Its main purpose is to study all the proteins in a complex mixture.
Experiment Process
The protein mixture in the venom is digested into a peptide mixture. After chromatographic separation and ionization of the peptide mixture, a peptide fingerprint is generated by tandem mass spectrometry for peptide identification. The data obtained is used to determine the original protein composition of the sample. With the improvement of mass resolution, mass accuracy, fragmentation technology and speed, bottom-up analysis methods can identify more proteins in complex samples than ever before.
Protein separation and purification
The complexity of the composition of venom samples from different species varies. The choice of the most appropriate sample preparation technique depends on the source, type, and complexity of the sample. Research goal may be to analyze a single purified protein, or it may classify thousands of proteins in a sample.
Preparation of peptides
The preparation of peptides involves reduction and alkylation of cysteine, digestion of samples into peptides, and desalting and concentration of peptides. Trypsin is by far the most commonly used digestive enzyme. When larger peptides are needed or the target region in the protein cannot produce suitable peptides, other enzymes such as GluC, AspN and LysC are sometimes used.
Data analysis
The optimal parameters of the MS instrument depend on the instrument type and sample complexity. There are a variety of algorithms for peptide fragmentation data analysis. We provide complete analysis tools and detailed information on data analysis.
Technology Platforms
- Liquid Chromatography (LC)
- High Performance Liquid Chromatography (HPLC)
- Time-of-flight mass spectrometry (MALDI-MS)
- Electrospray ionization mass spectrometry (ESI-MS)
One-stop Services
Creative Proteomics provides you with a one-stop service for protein full spectrum analysis, including sample processing, computer analysis, data analysis, and project reports. You only need to place an order and send the sample. We provide you with detailed experimental information, including:
- Experimental steps
- Related mass spectrometry parameters
- Mass spectrum picture
- Raw data
- Details of the identified protein
Advantages
- Can realize large-scale protein identification and quantification
- Comparable analysis of different samples
- Ideal for discovering and researching protein biomarkers
- Can be applied to dynamic proteomics analysis
Our protein full spectrum analysis services, using ultra-high resolution mass spectrometry technology, can help you comprehensively identify a large number of proteins in complex mixed samples.
Please contact us for more information.
For research use only. Not intended for any clinical use.