What is Co-IP Mass Spec?
When it comes to identifying and understanding intricate protein interactions, there's a groundbreaking technique that's taking the scientific community by storm: Co-immunoprecipitation Mass Spectrometry (Co-IP/MS). Co-immunoprecipitation (Co-IP) is a widely used method for investigating protein-protein interactions in a complex mixture. However, combining Co-IP with mass spectrometry (MS) analysis has revolutionized the way researchers identify and characterize protein interactions. This advanced technique, known as Co-IP mass spectrometry (Co-IP-MS), offers an unprecedented level of resolution, sensitivity, and accuracy in detecting even weak protein interactions.
Learn more
Principles of Co-IP Mass Spectrometry
Co-IP/MS marries two seemingly simple techniques to create an awe-inspiring investigative method. Co-IP selectively captures protein complexes, while MS analysis goes one step further, identifying and quantifying the proteins within these complexes. Together, Co-IP/MS offers a comprehensive lens into the web of protein interaction networks.
The central principle of Co-IP-MS involves the selective capture of protein complexes by specific antibodies, followed by the identification and quantification of individual proteins within these complexes using mass spectrometry. By analyzing multiple samples at once, it becomes possible to map out intricate protein interaction networks, revealing key players and hubs within each network.
Furthermore, Co-IP-MS is not limited to studying static protein-protein interactions. The technique also enables researchers to capture and identify dynamic protein complexes that change in response to cellular signaling or environmental cues. This flexibility opens up exciting possibilities for studying complex biological systems, such as protein interaction networks in disease contexts or the interplay between different proteins in cellular processes.
Overall, Co-IP-MS represents a powerful tool for unraveling the complex and dynamic protein-protein interactions that underlie many biological processes. By combining the selectivity of Co-IP with the sensitivity and resolution of mass spectrometry, this technique has the potential to advance our understanding of fundamental cellular mechanisms and the development of new therapeutic approaches.
Workflow of Co-IP mass spectrometry
Co-IP: The protein complexes of interest are immunoprecipitated using specific antibodies that target a particular protein within the complex. The antibody-protein complexes are captured on solid supports such as beads.
Washing and Elution: The captured complexes are extensively washed to remove non-specifically bound proteins and contaminants. The elution step then releases the proteins from the solid supports, generating a purified protein complex sample.
Protein Digestion: The eluted protein complex is subjected to enzymatic digestion, typically using trypsin. This breaks down the proteins into smaller peptide fragments.
Mass Spectrometry Analysis: The resulting peptides are analyzed using mass spectrometry. The peptides are ionized and separated based on their mass-to-charge ratios. The ionized peptides are then detected and quantified, providing information about the presence, identity, and abundance of proteins within the complex.
Data Analysis: The mass spectrometry data is analyzed using bioinformatics tools and databases to identify the proteins present in the complex. Comparative analysis can be performed to assess changes in protein composition between different samples or conditions.
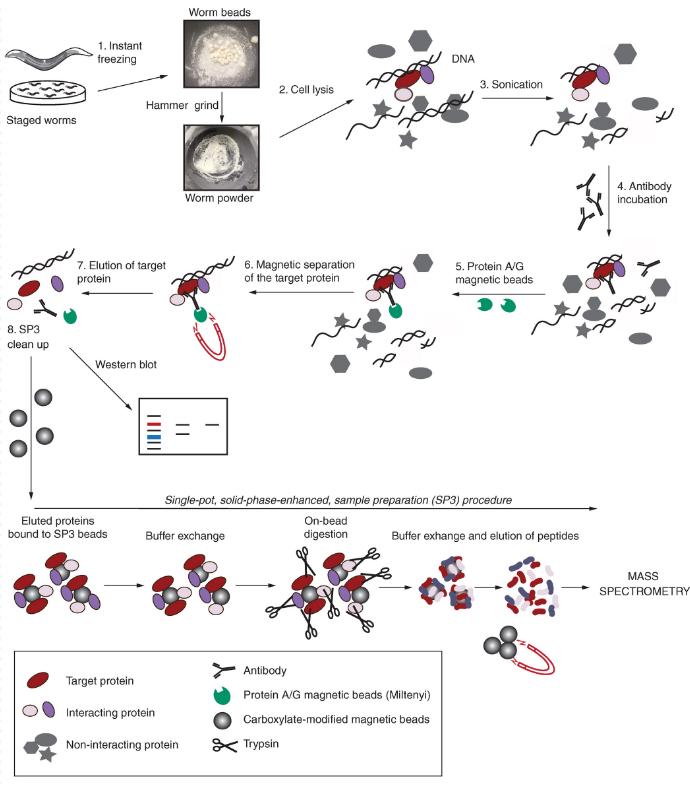
Co-IP/MS Technical Considerations and Optimization
Co-IP/MS its success depends on the careful optimization of various technical factors. This section will cover key considerations in the Co-IP-MS workflow, including antibody selection, buffer optimization, sample preparation, cross-linking strategies and data acquisition/analysis.
Antibody Selection and Specificity
The choice of antibody is one of the most critical factors in a Co-IP experiment. The antibody needs to specifically recognize and bind the target protein (bait), enabling the isolation of its interacting partners.
Antibody Specificity: Antibodies must be highly specific to the bait protein to ensure that the captured complexes are biologically relevant. Cross-reactivity with other proteins can lead to non-specific binding and false positives. Monoclonal antibodies, which bind to a single epitope, are often preferred for their high specificity, while polyclonal antibodies can sometimes yield broader binding and increased sensitivity, but may also lead to non-specific interactions.
Antibody Affinity: The affinity of the antibody to its target is important for the efficiency of the pull-down. High-affinity antibodies will bind the bait protein effectively, but may also cause over-capture of interacting proteins or non-specific binding. Conversely, low-affinity antibodies might fail to isolate sufficient quantities of protein complexes. Thus, balancing the affinity of the antibody is key.
Antibody Validation: To ensure the reliability of the results, antibodies should be validated for their specificity and functionality through various methods such as western blotting, immunofluorescence, or protein knockout studies. Commercial antibodies may come with documentation on their validation, but custom antibodies often require more rigorous validation steps.
Optimization of Lysis and Binding Buffers
The choice of lysis buffer is crucial for extracting proteins from cells while maintaining the integrity of protein-protein interactions. The buffer composition affects both the efficiency of protein extraction and the preservation of interactions.
Lysis Buffer Composition: Lysis buffers typically contain detergents to solubilize membranes and salts to maintain protein stability. The type and concentration of detergent (e.g., Triton X-100, NP-40, or CHAPS) should be optimized based on the cellular compartment targeted and the protein solubility. Harsh detergents can disrupt protein complexes, while mild detergents might not extract sufficient amounts of protein.
Protease and Phosphatase Inhibitors: Protease inhibitors (e.g., PMSF, aprotinin, leupeptin) should always be included in the lysis buffer to prevent the degradation of proteins during cell disruption. Additionally, phosphatase inhibitors (e.g., sodium orthovanadate) are essential if the goal is to study protein interactions involving phosphorylation, as these inhibitors prevent dephosphorylation during sample preparation.
Buffer Ionic Strength and pH: The ionic strength and pH of the lysis buffer should be optimized to match the biological conditions under which the protein-protein interactions occur. A buffer with moderate salt concentrations (e.g., 150 mM NaCl) is often used to mimic physiological conditions, but in some cases, high-salt buffers may be necessary to isolate specific protein complexes or to reduce non-specific binding. The pH should be adjusted according to the pI of the target protein to maintain its solubility and functionality.
Washing Conditions to Minimize Non-Specific Binding
After the bait protein and its interactors have been captured by the antibody, it is essential to wash the beads thoroughly to remove non-specifically bound proteins. However, overly stringent washing can disrupt weak or transient interactions, while insufficient washing can lead to contamination by non-specific proteins.
Stringency of Washing Buffers: The washing conditions should be optimized to balance between removing non-specifically bound proteins and preserving the target protein-protein interactions. Typically, low salt or high salt conditions are tested to fine-tune specificity. The addition of detergents like Triton X-100 or NP-40 to washing buffers can help reduce non-specific interactions without disrupting strong protein-protein interactions.
Multiple Wash Steps: To improve specificity, multiple rounds of washing with increasing salt concentrations can be applied. The first wash removes the majority of non-specifically bound proteins, while subsequent washes help to refine the interaction profile by removing less tightly associated proteins.
Duration of Washing: The duration of the washing steps should also be optimized. Prolonged washing times can reduce the amount of bound protein, while short washing steps may allow contaminants to remain in the sample. Optimizing the time and temperature conditions is important to ensure that the protein complexes are stable during the washing process.
Cross-Linking to Stabilize Transient Interactions
Transient or weak protein-protein interactions, which are often difficult to capture, can be stabilized through chemical cross-linking. Cross-linkers covalently link interacting proteins, enabling the identification of even short-lived complexes.
Types of Cross-Linkers: Several cross-linking agents are available, with varying spacer lengths and reactivity. Popular cross-linkers include formaldehyde, which forms reversible bonds, and the amine-reactive cross-linkers like DSP (dithiobis-succinimidyl propionate), which stabilize protein complexes. The choice of cross-linker depends on the nature of the interactions being studied and the proteins' amino acid composition.
Optimization of Cross-Linking Conditions: The concentration of cross-linker, reaction time, and temperature need to be carefully optimized to ensure that cross-linking is effective without over-cross-linking, which could result in a complex that is too large to be analyzed by MS. Cross-linking should also be carried out at physiological pH and temperature to preserve the natural interactions.
Cross-Linking Reversal: After Co-IP, cross-linking reagents often need to be removed or reversed to avoid interference with downstream analysis, especially mass spectrometry. Specific reagents (e.g., dithiothreitol, tris(2-carboxyethyl)phosphine) are used to cleave cross-links without disrupting protein integrity.
Mass Spectrometry Data Acquisition and Analysis
After successful immunoprecipitation, the isolated protein complexes are analyzed using mass spectrometry. However, the quality of the Co-IP-MS results heavily relies on the optimization of data acquisition and post-analysis steps.
MS Instrumentation: The choice of mass spectrometer (e.g., Orbitrap, Q-TOF, or ion trap) and the mode of acquisition (e.g., data-dependent acquisition vs. data-independent acquisition) can influence the sensitivity, accuracy, and depth of the analysis. High-resolution and high-accuracy instruments are often preferred for identifying low-abundance proteins in complex mixtures.
Peptide Fractionation and Enrichment: To improve the coverage of the interactome, peptide fractionation techniques (e.g., SDS-PAGE, strong cation exchange chromatography) can be employed to separate complex samples into smaller fractions, thereby enhancing the number of identifications. This step is particularly important for low-abundance interacting partners.
Data Analysis and Validation: The resulting mass spectrometry data must be analyzed with specialized software (e.g., MaxQuant, Proteome Discoverer) to identify proteins and protein complexes. Bioinformatic tools can help filter out non-specific proteins and contaminants. Additionally, validation strategies, such as comparing Co-IP results with public protein interaction databases or performing orthogonal validation via western blotting, should be employed to confirm the accuracy of the findings.
Quantification Approaches: Label-free quantification (LFQ), stable isotope labeling, or Tandem Mass Tag (TMT) approaches can be used to quantify the abundance of interacting proteins. These methods allow for the comparison of protein complex composition under different experimental conditions, such as drug treatments, stress responses, or disease models.
Quality Control and Reproducibility
Ensuring the reliability and reproducibility of Co-IP-MS experiments is critical for obtaining meaningful biological insights.
Negative Controls: Including appropriate controls, such as using an isotype control antibody or a bait protein that is known not to interact with other proteins, is essential for assessing the specificity of the Co-IP experiment and eliminating non-specific binding.
Replication and Statistical Analysis: Replicating Co-IP experiments under the same conditions (technical replicates) or using biological samples from independent experiments (biological replicates) helps to assess the consistency and reliability of the results. Statistical analysis can be used to determine the significance of identified interactions, and computational approaches can help evaluate the robustness of the interaction data.
Applications of Co-IP Mass Spectrometry in Protein Interactions
Co-IP coupled with MS can identify and characterize proteins that interact within cellular complexes, revealing how these interactions control cellular processes and contribute to disease mechanisms.
Protein Complex Identification
One of the primary uses of Co-IP coupled with mass spectrometry is the identification of proteins within specific complexes. Traditional methods of protein complex analysis, such as affinity purification followed by gel electrophoresis, can provide basic insights, but Co-IP-MS offers a more detailed and high-throughput approach.
- Co-IP Strategy: In this approach, an antibody specific to a bait protein is used to isolate the protein complex in which the bait is involved. The proteins that co-precipitate with the bait are then identified by MS. The use of high-resolution mass spectrometers allows for the precise identification of not only the bait protein but also the other interacting partners.
- Applications in Complex Composition: This method provides insights into the composition of large macromolecular complexes, including multi-subunit complexes involved in processes like signal transduction, transcriptional regulation, and cellular trafficking. For example, Co-IP-MS has been used to identify components of the spliceosome, chromatin remodeling complexes, and ribosomal subunits.
- Condition-Specific Complexes: By comparing Co-IP-MS data under different conditions (e.g., before and after a cellular stress response or drug treatment), researchers can identify condition-specific changes in complex composition, uncovering regulatory mechanisms or identifying novel protein complexes involved in cellular adaptation.
Protein Interaction Network Mapping
Co-IP mass spectrometry is a powerful tool for constructing comprehensive protein interaction networks (PINs). These networks map how proteins physically interact and can reveal functional relationships that are often not apparent from genetic or expression data alone.
- Network Construction: By performing Co-IP experiments with a variety of baits (e.g., proteins known to function in the same pathway or biological process), researchers can build a comprehensive interaction map. Mass spectrometry allows the simultaneous identification of hundreds or even thousands of interacting proteins from a single pull-down experiment, contributing to the creation of robust protein interaction networks.
- Applications in System Biology: These interaction maps are essential for systems biology approaches, where the goal is to understand the behavior of the entire proteome or specific cellular pathways. The networks can be used to identify hubs (central proteins with many interactions), modules (groups of interacting proteins), or key regulators that control cellular functions. Additionally, they can be instrumental in drug target discovery by identifying potential protein targets or protein inhibitors that disrupt critical interactions.
- Pathway Discovery and Functional Annotation: Interaction networks derived from Co-IP-MS data can be analyzed using bioinformatics tools to assign functional annotations to proteins based on their interaction profiles. This is particularly valuable for poorly characterized or newly discovered proteins.
Detection of Transient Interactions
Transient protein interactions are often difficult to study due to their brief and weak nature. These interactions play crucial roles in processes like signal transduction, cellular response to stress, and cell cycle regulation. Traditional methods like yeast two-hybrid or affinity chromatography are not well-suited to detect such fleeting interactions, but Co-IP-MS can provide a solution.
- Optimizing Experimental Conditions: The key to detecting transient interactions is optimizing experimental conditions, such as using mild washing buffers to retain weak interactions or employing cross-linking agents to stabilize transient complexes. Cross-linking agents like formaldehyde or DSP (dithiobis-succinimidyl propionate) can stabilize protein interactions in vivo, allowing for the capture of transient complexes before they dissociate.
- Dynamic Interactions and Signaling: Co-IP-MS is used extensively in the study of signaling pathways, where transient protein-protein interactions occur rapidly in response to cellular stimuli. For example, the binding of a ligand to a receptor often triggers a cascade of transient interactions within seconds to minutes. By applying Co-IP-MS under different time points after ligand treatment, researchers can track these dynamic events and identify key signaling intermediates.
Protein Interaction Quantification
Quantifying the interactions between proteins is crucial for understanding the stoichiometry, affinity, and dynamics of protein complexes. Co-IP-MS offers the ability to quantify not only the presence of proteins in a complex but also the relative abundance of each component. This can provide insights into how protein interactions change under different physiological or experimental conditions.
- Label-Free Quantification (LFQ): In label-free quantification, the intensity of peptide peaks in the mass spectrum is used to estimate the relative abundance of proteins in different Co-IP samples. By comparing samples obtained under various conditions (e.g., in the presence or absence of a drug), researchers can assess changes in protein complex composition or interaction strength.
- Isotope Labeling Approaches: Alternatively, stable isotope labeling methods such as SILAC (Stable Isotope Labeling by Amino Acids in Cell Culture) or TMT (Tandem Mass Tags) can be used to introduce isotopic tags into the samples, allowing for more accurate quantification across different experimental conditions. For instance, changes in the abundance of a protein partner in a complex can indicate altered interaction dynamics or changes in protein complex assembly.
- Studying Interaction Strength and Stoichiometry: Co-IP-MS can also be used to assess the stoichiometry of interacting proteins. The ratio of proteins in a complex can be quantified, revealing whether a protein exists in a 1:1 ratio with its partner or in a more complex, multimeric arrangement. This information is particularly important in understanding the functional architecture of complexes and their regulatory mechanisms.
Investigating Disease Mechanisms
Co-IP-MS has proven instrumental in investigating the molecular basis of diseases, particularly those related to dysfunctional protein-protein interactions. Many diseases, including cancer, neurodegeneration, and viral infections, involve the disruption of normal protein complexes.
- Cancer Research: In cancer, altered PPIs often lead to the dysregulation of cell signaling pathways, leading to uncontrolled cell proliferation and survival. Co-IP-MS can identify aberrant protein interactions in cancer cells, offering potential biomarkers for diagnosis and new targets for therapeutic intervention.
- Neurodegenerative Diseases: In diseases such as Alzheimer's, Parkinson's, and Huntington's, the aggregation of misfolded proteins and the formation of aberrant protein complexes is a hallmark. Co-IP-MS can be used to study how disease-associated proteins interact with other cellular machinery, potentially identifying new therapeutic targets or biomarkers.
- Viral Infections: Viruses often hijack host cell machinery by forming complexes with host proteins. Co-IP-MS has been widely used to identify viral-host protein interactions, revealing how viruses like HIV, influenza, and SARS-CoV-2 manipulate host cell processes for replication. These studies can also highlight potential drug targets for antiviral therapies.
Significance in Understanding Complex Biological Processes
Co-immunoprecipitation combined with mass spectrometry has revolutionized our understanding of complex biological processes. It has shed light on intricate protein interaction networks, signaling pathways, and disease mechanisms. By identifying interacting partners and characterizing their associations, researchers can unravel the functional significance of protein interactions in various cellular processes, such as signal transduction, protein trafficking, and gene regulation.
Furthermore, Co-IP mass spectrometry can be utilized to identify novel interacting partners and validate known protein interactions. This information contributes to the discovery of potential therapeutic targets and the development of targeted therapies. By understanding the protein interactions involved in disease pathways, researchers can identify key molecules that can be targeted to modulate the aberrant protein-protein interactions and restore normal cellular functions.
The integration of Co-IP mass spectrometry with other omics approaches, such as genomics, transcriptomics, and proteomics, provides a comprehensive view of the molecular landscape and enables a systems-level understanding of biological processes. This multidimensional analysis allows for the identification of functional modules, protein complexes, and regulatory networks that contribute to the complexity of cellular functions.
In conclusion, Co-immunoprecipitation (Co-IP) combined with mass spectrometry is a powerful approach for studying protein interactions and unraveling complex biological processes. It provides valuable insights into protein complex composition, protein interaction networks, and dynamic changes in protein interactions. The integration of Co-IP with mass spectrometry enables the identification of interacting partners, quantification of protein interactions, and detection of transient interactions. This information has significant implications for understanding disease mechanisms, identifying therapeutic targets, and developing targeted therapies.
Creative Proteomics, a leading provider of proteomics services, offers expertise in Co-IP mass spectrometry analysis. With their state-of-the-art facilities and experienced scientists, they can assist researchers in unraveling intricate protein interaction networks and identifying novel therapeutic targets. Creative Proteomics employs cutting-edge mass spectrometry platforms and advanced data analysis tools to ensure accurate and reliable results.
With the expertise of Creative Proteomics, researchers can harness the full potential of Co-IP mass spectrometry to advance our knowledge of protein-protein interactions and accelerate biomedical research and drug discovery.
Reference
- Gülkiz Baytek et al,. Robust co-immunoprecipitation with mass spectrometry for Caenorhabditis elegans using solid-phase enhanced sample preparation BIOTECHNIQUES 2022