The TCA cycle functions as the central hub of aerobic metabolism, operating within mitochondrial matrices to oxidize acetyl-CoA derived from carbohydrates, lipids, and amino acids. Through this process, it generates ATP, NADH, and FADH₂—critical energy carriers that fuel cellular activities. Given its pivotal role in bioenergetics, the TCA cycle is tightly governed by multilayered regulatory mechanisms to balance ATP synthesis with metabolic stability. This review systematically examines four principal modes of cycle regulation: allosteric control, substrate accessibility, post-translational modifications, and transcriptional programming, which collectively ensure adaptability to fluctuating cellular demands.
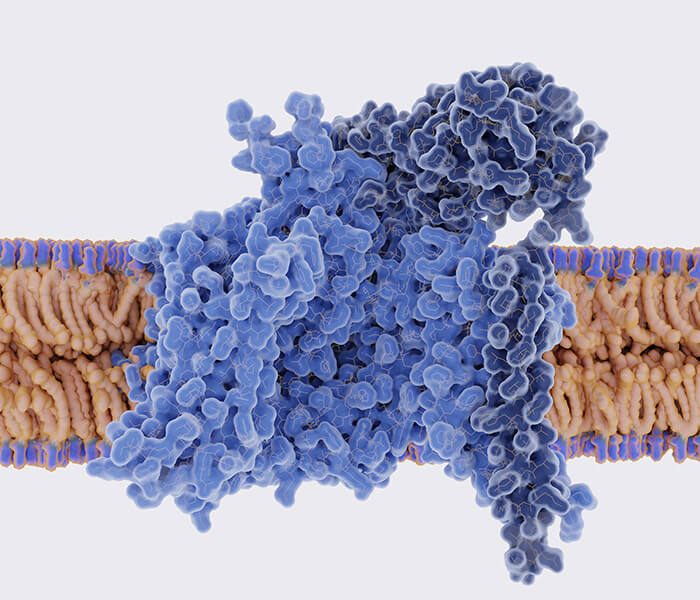
 TCA cycle produces reduction equivalents NADH and FADH2 (Martínez-Reyes I et al., 2020).
TCA cycle produces reduction equivalents NADH and FADH2 (Martínez-Reyes I et al., 2020).
Allosteric Regulation
Allosteric modulation is pivotal for dynamically adjusting the TCA cycle's flux, enabling cells to align metabolic activity with energy demands. By binding effector molecules to regulatory sites, enzymes undergo conformational changes that enhance or suppress catalytic activity, ensuring efficient energy utilization and preventing metabolic imbalance. This responsive mechanism allows rapid adaptation to fluctuations in cellular energy status, optimizing ATP synthesis while conserving resources.
Key Enzymes and Regulatory Mechanisms
Citrate Synthase
- Function: Catalyzes the condensation of acetyl-CoA and oxaloacetate to initiate the cycle by forming citrate.
- Regulation: Inhibited by ATP, NADH, and succinyl-CoA—molecules signaling energy surplus. This feedback inhibition prevents excessive cycle activity under energy-replete conditions, maintaining metabolic equilibrium.
Isocitrate Dehydrogenase (IDH)
- Function: Converts isocitrate to α-ketoglutarate, generating NADH and CO₂.
- Regulation:
- Activation: ADP (indicating low ATP) and Ca²⁺ (signaling elevated metabolic demand, e.g., muscle contraction) enhance activity.
- Inhibition: ATP and NADH suppress IDH during energy abundance, curbing cycle flux to avoid NADH overproduction.
α-Ketoglutarate Dehydrogenase (α-KGDH)
- Function: Oxidizes α-ketoglutarate to succinyl-CoA, a critical step linked to NADH generation.
- Regulation: Inhibited by succinyl-CoA and NADH. Their accumulation signals downstream metabolite sufficiency, ensuring cycle deceleration to prevent redundant energy expenditure.
Biological Significance
- Energy Efficiency: Allosteric controls prevent futile cycling, directing resources only when ATP demand rises (e.g., during stress or growth).
- Metabolic Flexibility: Enables swift transitions between anabolism and catabolism by modulating enzyme activity in response to cellular cues (e.g., ADP/ATP ratio, calcium signaling).
- Disease Relevance: Dysregulation of these mechanisms is implicated in pathologies such as cancer (e.g., IDH mutations) and mitochondrial disorders, highlighting their therapeutic potential.
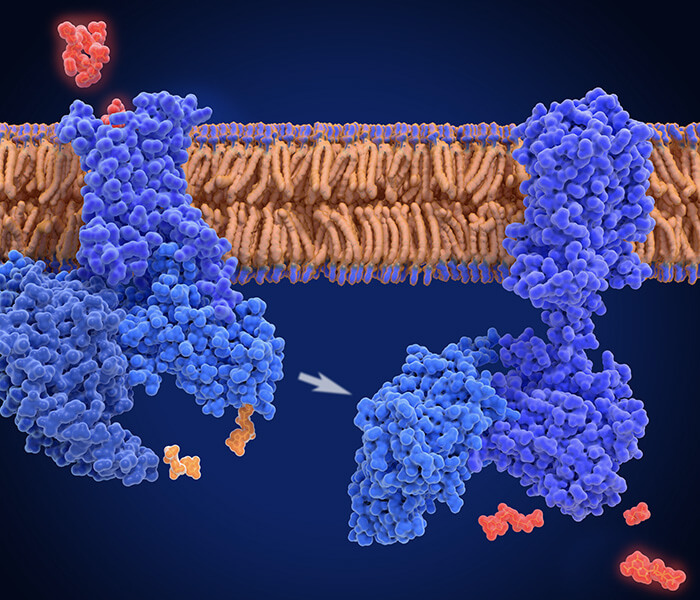
 Allosteric and covalent regulation of the PDHC complex (Arnold PK et al., 2023).
Allosteric and covalent regulation of the PDHC complex (Arnold PK et al., 2023).
Services You May Be Interested In
Additional Resources
Substrate availability
The TCA cycle is critically regulated by substrate accessibility. Its functionality and progression depend on the influx of metabolites like acetyl-CoA and oxaloacetate, whose concentrations, availability, and cellular demand collectively influence metabolic network dynamics.
Acetyl-CoA Availability
As a primary entry point into the TCA cycle, acetyl-CoA originates from various metabolic precursors. Its synthesis pathways include:
- Carbohydrate Catabolism: Glycolysis converts glucose to pyruvate, which is subsequently transformed into acetyl-CoA via the pyruvate dehydrogenase complex (PDH). Elevated glucose availability enhances acetyl-CoA production, stimulating TCA cycle activity.
- Lipid β-Oxidation: During energy-intensive states (e.g., fasting or exercise), fatty acids undergo β-oxidation to generate acetyl-CoA. This pathway becomes dominant under prolonged energy deficits, ensuring sustained cycle operation.
- Amino Acid Degradation: Branched-chain amino acids (e.g., leucine, isoleucine) contribute to acetyl-CoA pools through transamination and deamination, particularly during protein-rich metabolic conditions.
Regulatory Mechanisms
Acetyl-CoA levels modulate citrate synthase activity and downstream reactions. While abundant acetyl-CoA boosts cycle flux, excessive accumulation triggers feedback inhibition—for instance, suppressing glycolytic pathways to prevent energy surplus.
Oxaloacetate Dynamics
Oxaloacetate serves as the acetyl-CoA acceptor, initiating citrate synthesis. Its availability directly governs cycle continuity through two primary mechanisms:
- Anaplerotic Replenishment: Depleted oxaloacetate pools activate replenishment pathways. Pyruvate carboxylase catalyzes pyruvate-to-oxaloacetate conversion, ensuring substrate sufficiency during carbon abundance.
- Alternative Precursors: Aspartate transamination and lactate oxidation (via lactate dehydrogenase and pyruvate carboxylase) provide supplementary oxaloacetate, sustaining cycle function during hypoxia or intense metabolic activity.
Regulatory Implications
Low oxaloacetate concentrations impair citrate synthase activity, stalling the cycle. Anaplerotic reactions restore equilibrium, preventing metabolic interruption and maintaining energy homeostasis.
Covalent modifications
Covalent modifications are central to numerous cellular metabolic processes, particularly in modulating enzymes critical for energy homeostasis. This post-translational regulation—especially phosphorylation and dephosphorylation—exerts profound influence over the TCA cycle's efficiency and adaptability.
PDC Regulation
PDC bridges glycolysis and the TCA cycle by catalyzing pyruvate's irreversible conversion to acetyl-CoA, a pivotal entry point for mitochondrial energy production. Its activity is dynamically controlled via reversible phosphorylation, mediated by pyruvate dehydrogenase kinase (PDK) and phosphatase (PDP).
PDK
PDK phosphorylates specific residues (e.g., E1 subunit) on PDC, rendering it inactive. This suppression occurs under energy-replete conditions signaled by elevated ATP, NADH, or acetyl-CoA. By inhibiting PDC, PDK reduces acetyl-CoA synthesis, thereby curbing TCA cycle flux to align with cellular energy sufficiency.
PDP
Conversely, PDP reverses PDK-mediated inhibition by dephosphorylating PDC, reactivating pyruvate oxidation. PDP is stimulated by energy-depleting signals such as low ATP, high AMP, and elevated NAD+ levels, ensuring enhanced acetyl-CoA production during heightened metabolic demand.
Metabolic Feedback Loops
Beyond PDK/PDP interplay, multiple metabolites fine-tune PDC phosphorylation:
- Acetyl-CoA: Excess levels reinforce PDK activity, creating a negative feedback loop to limit further acetyl-CoA generation.
- NADH: Elevated concentrations amplify PDK signaling, reducing TCA cycle activity to prevent redox imbalance.
- ATP/AMP Ratios: High ATP stabilizes PDC's inactive state, whereas AMP accumulation promotes PDP-driven activation, synchronizing energy synthesis with demand.
Systemic Metabolic Integration
PDC phosphorylation status serves as a metabolic nexus, integrating energy status with broader cellular physiology:
- During fasting, low ATP and rising AMP levels prioritize PDP activity, maximizing acetyl-CoA synthesis to sustain energy output.
- In nutrient-rich states, PDK-mediated inhibition prevents wasteful substrate cycling, aligning TCA flux with biosynthetic and redox needs.
- This regulatory axis also underpins cellular stress adaptation, enabling rapid metabolic reprogramming during oxidative challenges or growth transitions.
Transcriptional control
The tricarboxylic acid cycle (TCA cycle, or citric acid cycle) is governed not only by enzymatic activity but also by transcriptional regulation, enabling long-term adaptation to cellular energy states, oxygen availability, and environmental cues. This dual-layered control involves transcription factors, signaling cascades, and metabolic sensors that modulate gene expression of TCA-associated enzymes. Below, we analyze key regulatory mechanisms shaping this critical metabolic pathway.
AMPK
As a central energy sensor, AMPK responds to low cellular ATP levels by monitoring the AMP/ATP ratio. Activation under energy stress triggers metabolic reprogramming to restore balance, prioritizing ATP synthesis while curbing non-essential processes.
AMPK's Regulatory Mechanisms:
- Mitochondrial Biogenesis: AMPK upregulates genes linked to oxidative phosphorylation (OXPHOS), enhancing mitochondrial proliferation and efficiency to bolster energy output.
- TCA Enzyme Modulation: By elevating pyruvate dehydrogenase kinase (PDK) expression, AMPK indirectly suppresses pyruvate dehydrogenase complex (PDC) activity, fine-tuning TCA flux to match energy demands.
- Fatty Acid Utilization: AMPK stimulates β-oxidation, increasing acetyl-CoA supply to fuel the TCA cycle during energy deficits.
PPARs
Members of the nuclear receptor superfamily (PPAR-α, PPAR-β/δ, PPAR-γ), PPARs coordinate lipid and glucose metabolism by binding fatty acid ligands. Their activation enhances transcriptional programs supporting energy production.
PPAR-Driven TCA Regulation:
- Lipid Metabolism Coordination: PPAR-α promotes fatty acid transport into mitochondria via carnitine palmitoyltransferase 1 (CPT1), amplifying acetyl-CoA generation for TCA cycle activation.
- Enzyme Upregulation: PPARs enhance expression of TCA enzymes (e.g., citrate synthase, isocitrate dehydrogenase) and OXPHOS components, optimizing cycle efficiency during sustained energy demands.
- Metabolic Flexibility: By prioritizing fatty acid oxidation, PPARs sustain TCA activity during fasting or prolonged exercise, aligning substrate utilization with physiological needs.
HIFs
HIFs stabilize under low oxygen, redirecting metabolism from oxidative pathways to glycolysis to conserve oxygen.
HIF-Mediated TCA Suppression:
- Glycolytic Shift: HIF-1α activates genes encoding glycolytic enzymes, reducing reliance on oxygen-dependent TCA cycle and OXPHOS.
- Enzyme Downregulation: HIFs repress citrate synthase and isocitrate dehydrogenase expression, attenuating TCA flux to minimize ROS generation under hypoxia.
- Anaerobic Prioritization: This metabolic reprogramming ensures ATP production via glycolysis, sustaining cell survival in oxygen-poor environments.
Auxiliary Transcriptional Regulators
Additional factors integrate stress, nutrient, and hormonal signals into TCA cycle modulation:
- SIRT1: This NAD+-dependent deacetylase activates PGC-1α, boosting mitochondrial biogenesis and TCA enzyme activity during caloric restriction or oxidative stress.
- FoxO Proteins: FoxO transcription factors adjust TCA-related gene expression in response to energy scarcity or oxidative damage, balancing metabolic output with cellular resilience.
- Thyroid Hormone Receptors (TRs): TRs modulate mitochondrial gene networks, influencing TCA cycle capacity in energy-intensive tissues.
Ca²⁺ signal
Ca²⁺ serve as critical signaling molecules in cellular processes, modulating energy homeostasis, mechanical contraction, and stress adaptation. Beyond their role in signal transduction, Ca²⁺ directly influences TCA cycle enzymes, synchronizing mitochondrial energy production with cellular demands. In myocytes, Ca²⁺ not only initiates contraction but also coordinates ATP synthesis by activating metabolic enzymes, ensuring energy supply meets physiological requirements. Below, we detail Ca²⁺'s mechanistic contributions to TCA cycle regulation and muscle function.
Calcium-Dependent Regulation of the TCA Cycle
Dynamic shifts in Ca²⁺ concentrations fine-tune TCA cycle activity through allosteric modulation of rate-limiting enzymes, particularly during high-energy states like muscle exertion.
Isocitrate Dehydrogenase (IDH) Activation
IDH catalyzes the oxidative decarboxylation of isocitrate to α-ketoglutarate, a NADH-generating step. Ca²⁺ binding induces conformational changes in IDH, augmenting its catalytic efficiency.
- Metabolic Synchronization: Elevated cytosolic Ca²⁺ during muscle activity accelerates IDH-driven NADH synthesis, sustaining ATP production to match contraction intensity.
- Adaptive Response: Transient Ca²⁺ spikes during exercise enhance carbon flux through the TCA cycle, preventing energy depletion during prolonged activity.
α-Ketoglutarate Dehydrogenase (α-KGDH) Enhancement
The α-KGDH complex facilitates the conversion of α-ketoglutarate to succinyl-CoA. Ca²⁺ potentiates this reaction by stabilizing enzyme-coenzyme interactions.
- Kinetic Amplification: Ca²⁺ increases α-KGDH's turnover rate, particularly under metabolic stress, ensuring rapid succinyl-CoA production for ATP and heme biosynthesis.
- Demand-Driven Regulation: During acute energy crises, Ca²⁺-mediated α-KGDH activation prioritizes substrate oxidation over anaplerotic pathways.
Calcium Signaling in Muscle Energetics
In skeletal and cardiac muscle, Ca²⁺ synchronizes mechanical activity with metabolic output, coupling contraction to mitochondrial respiration.
Excitation-Contraction Coupling
Neuronal stimulation triggers Ca²⁺ release from the sarcoplasmic reticulum, initiating sarcomere shortening via actin-myosin cross-bridge cycling. Concurrently, cytosolic Ca²⁺ surges activate Ca²⁺/calmodulin-dependent kinases, priming metabolic pathways.
Metabolic-Contractile Coupling
- ATP Synthesis Optimization: Ca²⁺ upregulates TCA flux by 30–50% during contraction, driven by IDH and α-KGDH activation, to replenish ATP consumed by myosin ATPases.
- Feedback Coordination: Mitochondrial Ca²⁺ uptake stimulates pyruvate dehydrogenase and ATP synthase, creating a feedforward loop that sustains contraction endurance.
Integrated Physiological Implications
- Exercise Adaptation: Repeated Ca²⁺ transients during training enhance mitochondrial biogenesis, amplifying TCA capacity over time.
- Pathological Contexts: Dysregulated Ca²⁺ handling in heart failure disrupts TCA enzyme activity, contributing to energetic insufficiency.
Redox equilibrium
The TCA cycle serves dual roles in cellular physiology: it is central to ATP synthesis and acts as a linchpin in maintaining redox equilibrium. This balance between oxidation and reduction states is vital for cellular survival, as deviations disrupt metabolic homeostasis and trigger dysfunction. Below, we explore how the TCA cycle intersects with redox regulation through NADH/NAD⁺ dynamics, ROS interactions, and antioxidant defenses.
NADH/NAD⁺ Ratio: A Metabolic Rheostat
The TCA cycle generates NADH via substrate oxidation (e.g., isocitrate, α-ketoglutarate), which transfers electrons to the electron transport chain (ETC) for ATP synthesis. The NADH/NAD⁺ ratio reflects cellular energy status and directly modulates TCA flux.
Elevated NAD⁺/NADH Ratio
A high NADH/NAD⁺ ratio signals energy surplus, prompting metabolic slowdown:
- Enzymatic Inhibition: Excess NADH allosterically suppresses TCA enzymes (e.g., isocitrate dehydrogenase, α-ketoglutarate dehydrogenase), curbing substrate oxidation to prevent NADH overproduction.
- ETC Saturation: When NAD⁺ becomes limiting, electron transfer stalls, reducing proton gradient efficiency and ATP synthesis. This feedback loop prevents energy waste under replete conditions.
Depleted NADH/NAD⁺ Ratio
Low NADH/NAD⁺ levels indicate energy demand, stimulating TCA activity:
- Cycle Acceleration: Increased NAD⁺ availability enhances enzyme kinetics, driving NADH and FADH₂ synthesis to fuel ATP generation.
- ETC Optimization: Elevated electron acceptor capacity ensures efficient oxidative phosphorylation, aligning energy output with cellular needs.
ROS: Dual Roles in Redox Stress
ROS, primarily mitochondrial byproducts, regulate redox signaling at physiological levels but induce oxidative damage when overproduced.
TCA Cycle Impairment by ROS
- Enzyme Inactivation: ROS oxidize cysteine residues in TCA enzymes (e.g., aconitase, α-KGDH), impairing catalytic activity and slowing carbon flux.
- Energy Crisis: Inhibited TCA function reduces NADH output, compromising ATP synthesis and exacerbating metabolic stress during oxidative injury.
ROS Generation and Mitigation
- Mitochondrial Leakage: Electron escape at ETC complexes I and III generates superoxide (O₂⁻), particularly under NADH overload or impaired ETC function.
- Antioxidant Buffering: Cells deploy glutathione peroxidase and superoxide dismutase to neutralize ROS, while thioredoxin systems repair oxidized enzymes, restoring TCA activity.
Integrated Redox Homeostasis
- Adaptive Responses: Low ROS levels activate Nrf2 signaling, upregulating antioxidant genes to bolster defenses. Conversely, chronic oxidative stress downregulates TCA enzymes to limit further ROS production.
- Metabolic Flexibility: The TCA cycle dynamically adjusts NADH/NAD⁺ ratios and ROS scavenging to balance energy synthesis with redox stability, ensuring resilience across physiological states.
References
- Arnold PK, Finley LWS. "Regulation and function of the mammalian tricarboxylic acid cycle." J Biol Chem. 2023 Feb;299(2):102838. doi: 10.1016/j.jbc.2022.102838
- Lian WS, Wu RW, Lin YH, Chen YS, Jahr H, Wang FS. "Tricarboxylic Acid Cycle Regulation of Metabolic Program, Redox System, and Epigenetic Remodeling for Bone Health and Disease." Antioxidants (Basel). 2024 Apr 17;13(4):470. doi: 10.3390/antiox13040470