Methyl-Proteomics Service
Creative Proteomics offers comprehensive protein methylation analysis services, utilizing advanced methyl-peptide enrichment techniques and high-resolution HPLC-MS/MS platforms. These services enable the precise identification and quantification of methylation sites, aiding research in epigenetics, cellular signaling, and disease mechanisms. Tailored workflows, including sample preparation, site-specific mapping, and quantitative analysis, ensure high specificity and reproducibility for various sample types, supporting studies in both basic research and therapeutic development.
Submit Your Request Now
×- Define
- What We Provide
- Advantages
- Sample Requirements
- Demo
- FAQs
- Case Study
- Publications
What is Protein Methylation?
Protein methylation is a type of post-translational modification that frequently occurs on specific methylated amino acids, notably arginine methylation and lysine methylation. This modification involves the addition of one or more methyl groups to the side chains of amino acids within a protein, which influences the protein's function and interaction with other molecules. Protein methylation is often catalyzed by specific enzymes that utilize S-Adenosylmethionine (SAM) as a methyl donor. For instance, at the lysine (Lys) site, the ε-amino group of Lys can undergo mono-, di-, or trimethylation catalyzed by lysine methyltransferases (KMTs). Meanwhile, arginine methylation occurs on arginine (Arg) residues, where protein Arg methyltransferases (PRMTs) can facilitate mono-, symmetric di-, or asymmetric di-methylation, also in a SAM-dependent manner.
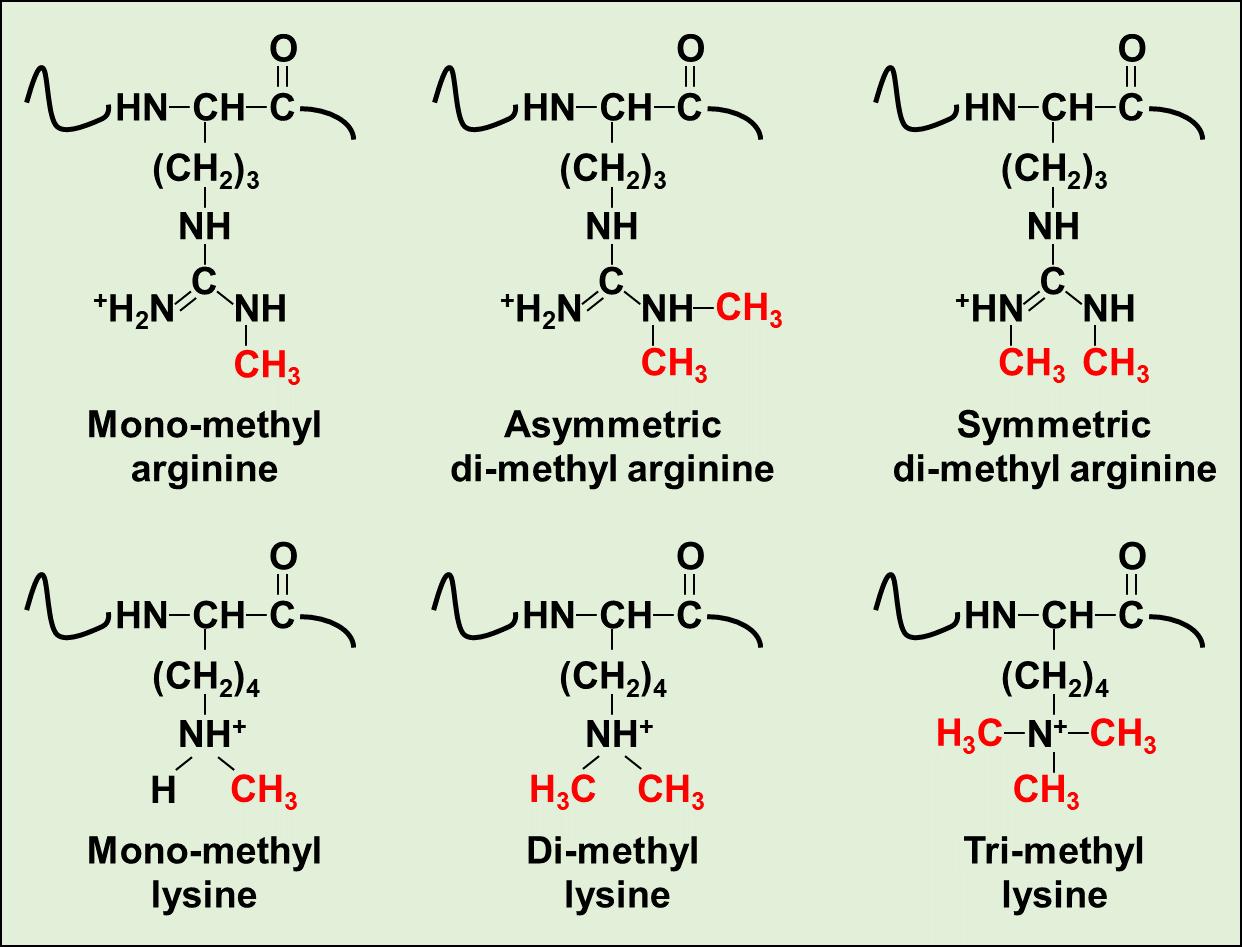 Figure 1. Chemical structures of six distinct methylated modification on Arg or Lys residues.
Figure 1. Chemical structures of six distinct methylated modification on Arg or Lys residues.
Protein methylation is significant in both histone and non-histone proteins, such as transcription factors, affecting various epigenetic and cellular processes. In histones, methylation, along with DNA methylation, constitutes an important epigenetic regulatory mechanism that influences gene expression. Additionally, protein methylation plays critical roles in cellular activities, including signal transduction, transcriptional regulation, DNA repair, gene activation, gene repression, RNA processing, and protein stability. The disruption or deregulation of amino acid methylation has been implicated in several human diseases, such as metabolic, neurodegenerative, and muscular disorders, underscoring the necessity of understanding methylation mechanisms to advance treatments.
However, global profiling of methylated sites within cells remains a challenging task. Key difficulties include the low abundance and low stoichiometry of methylated proteins, the lack of effective biochemical techniques for enriching methylated peptides, and the complication of isobaric amino acid substitutions that resemble methylation. Therefore, gaining deeper insights into protein methylation is essential to unravel complex biological processes and improve therapeutic approaches.
Our Methyl-proteomics Service
Creative Proteomics has established advanced strategies for highly specific enrichment of methyl (Lysine)-peptides or methyl (Arginine)-peptides, along with a sensitive HPLC-MS/MS pipeline capable of analyzing various types of methylation in both eukaryotic and prokaryotic organisms. In addition, the methyl-proteomics service protocol has been optimized, facilitating a more efficient and precise site mapping for methyl-sites. Classically, the workflow of this service includes protein isolation from biological samples, proteolytic digestion into peptides, methyl-peptide enrichment utilizing immunoaffinity, hydrophilic interaction chromatography (HILIC), or Strong Cation exchange (SCX), identification of methylation sites, and comprehensive quantification of relative abundance changes for methylation sites. Additionally, the application of LC-MS/MS analysis can be complemented by incorporating iTRAQ/TMT labeling or SILAC labeling techniques to enhance the precision of relative quantitation between samples.
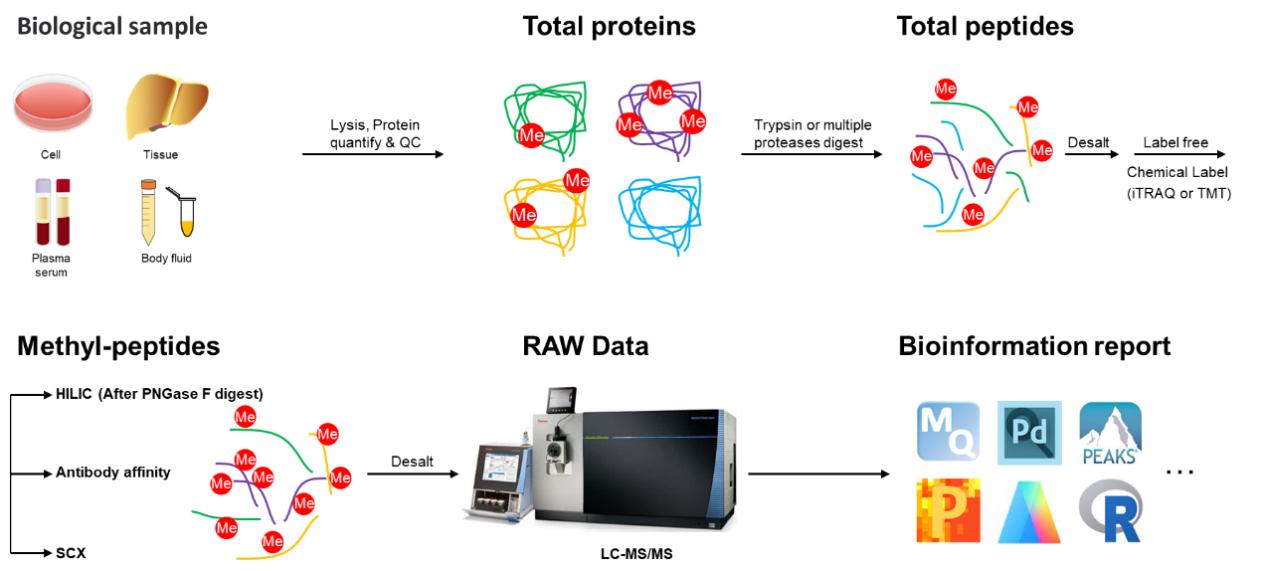 Figure 2. General Workflow for Methyl-proteomics Analysis.
Figure 2. General Workflow for Methyl-proteomics Analysis.
Technological Superiority
- Professional detection and analysis capability: Experienced PTM research team, strict quality control system, together with ultra-high resolution detection system and professional data pre-processing and analysis capability, ensure reliable and accurate data.
- Reproducible: Obtain consistent and reproducible inter- and intra- assay results for data analysis.
- High specificity: Use methylation-specific antibodies for methyl-peptides enrichment.
- Multiplex, high-throughput: Deeper coverage of methylation site identification.
- High resolution and sensitivity: Q-Exactive, Q-Exactive HF, Orbitrap Fusion™ Tribrid™.
Sample Requirements for Protein Methylation Analysis
| Sample Type | Recommended Sample Amount |
|---|---|
| Cell samples | 107 - 108 cells |
| Tissue samples | 100–200 mg of fresh or frozen tissue |
| Serum/Plasma | Minimum of 500 µL |
| Purified protein | Minimum of 50–100 µg |
| Cerebrospinal fluid (CSF) | Minimum of 1 mL |
| Urine | Minimum of 10 mL |
| Saliva | Minimum of 1 mL |
| Peripheral Blood Mononuclear Cells (PBMCs) | 5–10 million cells |
| Bone marrow | Minimum of 1 mL aspirate |
| Plant tissue | 100–200 mg of fresh or frozen tissue |
| Microbial cultures | 1–5 mL of culture at OD600 of 0.5–1.0 |
Demo
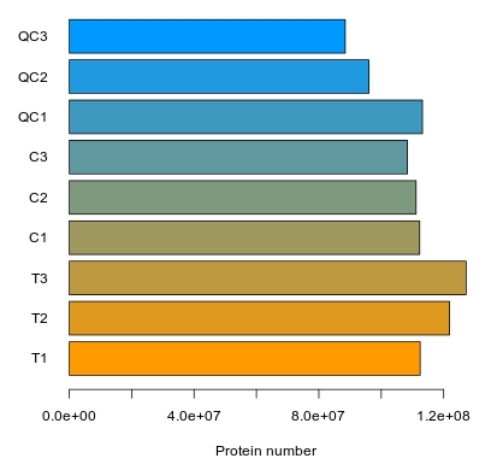
Bar Chart of Total Protein Identification
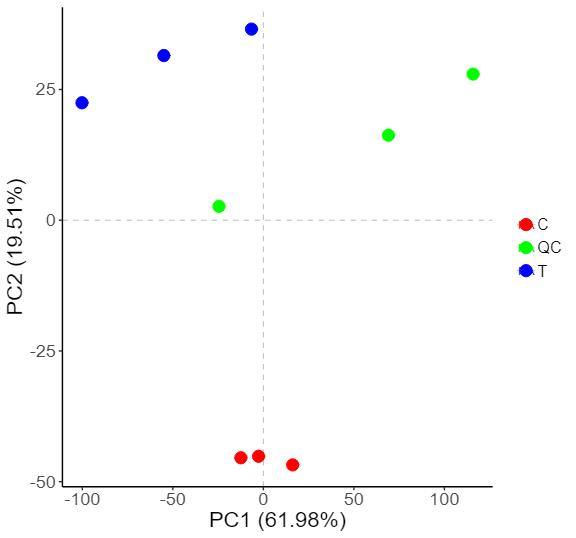
2D PCA Plot of Sample Grouping
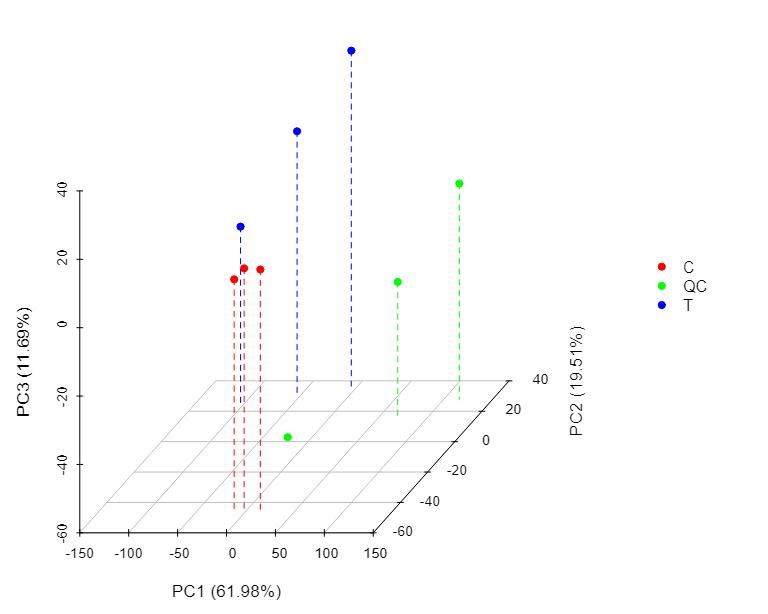
3D PCA Plot of Sample Grouping
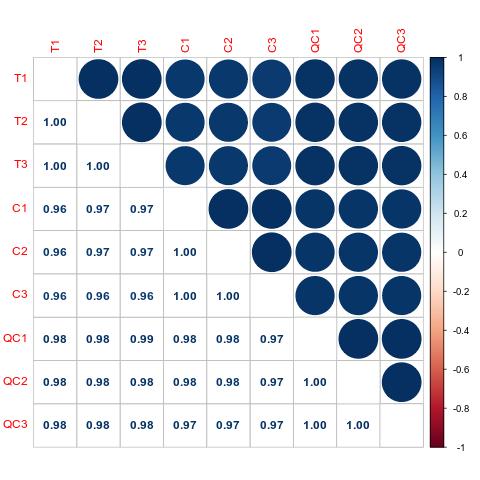
Pearson Correlation Analysis
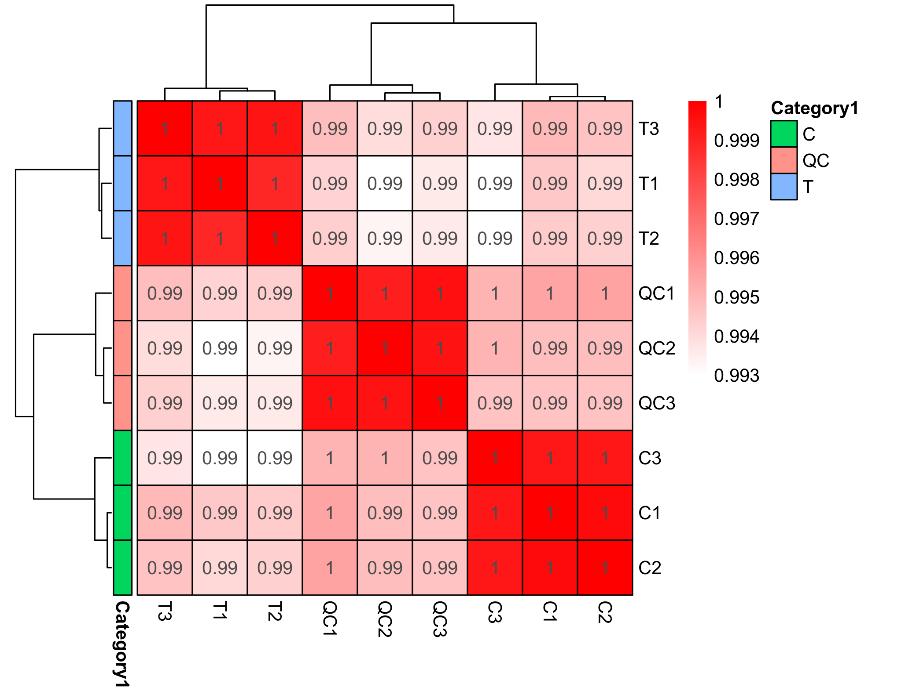
Sample Hierarchical Clustering
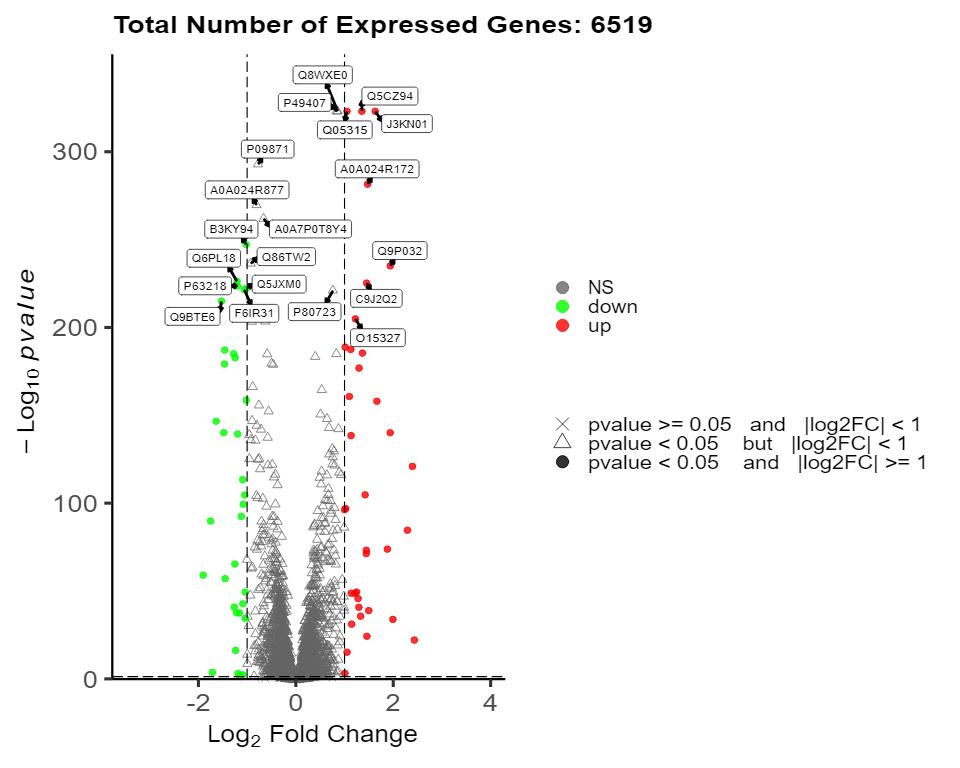
Volcano Plot of Differential Proteins
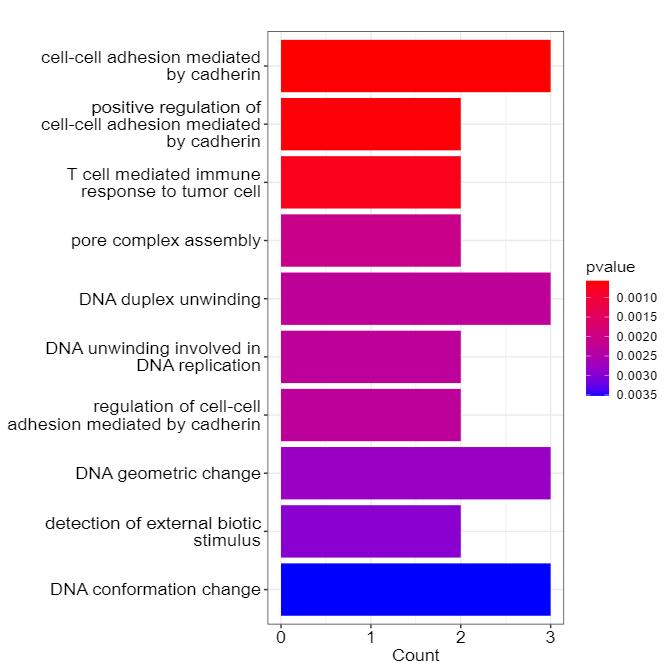
Bar Chart of GO Enrichment for Candidate Proteins
KEGG Pathway Enrichment of Candidate Proteins
FAQ
What factors should I consider when preparing my samples for methyl-proteomics analysis?
When preparing samples, it's crucial to ensure purity and optimal concentration. Biological matrices, such as tissue or cell samples, should ideally be free from contaminants like nucleic acids, polysaccharides, and lipids, which can interfere with peptide separation and MS/MS detection. Samples should also avoid substances like SDS, thiourea, or strong ion salts unless you specify their presence, as these can impact ionization efficiency and downstream analysis. If you're submitting protein samples, check the buffer to ensure compatibility with the HPLC-MS/MS setup.
How does Creative Proteomics ensure high specificity in methyl-peptide enrichment?
Creative Proteomics employs methylation-specific antibodies alongside techniques such as hydrophilic interaction chromatography (HILIC) and strong cation exchange (SCX) to selectively enrich methylated peptides. This multi-method approach minimizes non-specific binding, ensuring that enriched peptides truly reflect methylation events. Additionally, our antibodies are rigorously validated to detect methylated lysine or arginine residues with high specificity, thus enhancing the reliability of methylation site identification.
What is the typical detection sensitivity for methylated peptides, and how can it be optimized?
Our Q-Exactive and Orbitrap Fusion™ Tribrid™ systems provide ultra-high sensitivity, often detecting methylated peptides at the femtomole level. To enhance detection sensitivity, especially for low-abundance methylation sites, we recommend sample concentration adjustments based on initial test results. The incorporation of labeling techniques like SILAC or TMT/iTRAQ can also improve quantitation sensitivity, providing more robust data on differential methylation across samples.
Are there specific challenges in methylation profiling of non-histone proteins?
Yes, non-histone protein methylation analysis can be challenging due to the vast diversity of these proteins and the lower abundance of methylation events compared to histones. Our service employs a refined enrichment process and high-resolution mass spectrometry to detect these low-abundance modifications. Additionally, using advanced data processing tools, we optimize the detection of subtle methylation events on transcription factors and other regulatory proteins, which may play crucial roles in cellular processes.
Can methyl-proteomics detect multiple types of methylation in the same sample?
Absolutely. Our HPLC-MS/MS pipeline and advanced MS methods allow for the identification of mono-, di-, and trimethylation on both arginine and lysine residues within the same sample. By using immunoaffinity enrichment combined with high-resolution mass spectrometry, we can map various methylation states accurately, even within complex biological samples.
What experimental controls do you recommend for quantitative methyl-proteomics?
For reliable quantification, we recommend including several controls: 1) non-methylated or baseline samples to identify methylation-specific signals, 2) methylated peptide standards to calibrate the MS response and quantify relative abundance, and 3) technical replicates to ensure data reproducibility. These controls are essential for interpreting differential methylation data accurately and minimizing experimental variability.
How are data analysis and interpretation handled for complex methyl-proteomics data?
Our bioinformatics team uses advanced software tools to identify and quantify methylation sites, map peptides to specific residues, and analyze patterns across experimental conditions. For complex datasets, we employ normalization and correction methods to handle variability and statistical analysis techniques to identify significant methylation changes. We also offer optional consulting to help interpret results, understand potential biological implications, and suggest follow-up experiments.
How is sample stability managed to prevent methylation changes during storage or transport?
Methylation can be affected by environmental factors, so we recommend freezing samples immediately after collection and storing them at -80°C to maintain stability. Avoid freeze-thaw cycles, as these can degrade methylated peptides. During transport, ensure samples remain on dry ice. For long-term storage or sensitive samples, lyophilization may be considered to protect methylation integrity.
What are the limitations in detecting low-abundance methylation sites, and how can they be addressed?
Low-abundance methylation sites can be challenging to detect due to low stoichiometry and background noise. Our enrichment techniques, including the use of high-specificity antibodies, increase the chances of detecting these rare modifications. For highly sensitive detection, we also recommend using SILAC or TMT labeling to boost the signal for low-abundance methylation events across different conditions.
Can Creative Proteomics help design experiments for specific methylation questions or hypotheses?
Yes, our scientists can provide support in designing experiments tailored to specific research questions. This includes advising on sample preparation, selecting appropriate enrichment and labeling methods, and choosing optimal MS settings based on the type of methylation or target protein of interest. By understanding your research goals, we can help you obtain precise, relevant data to answer specific methylation-related hypotheses.
Learn about other Q&A.
Case Study

Deep Protein Methylation Profiling by Combined Chemical and Immunoaffinity Approaches Reveals Novel PRMT1 Targets.
Journal: Molecular & Cellular Proteomics
Published: 2019
- Highlights
- Background
- Main Technology
- Results
1. Enrichment of methyl peptides using two orthogonal techniques.
2. Knockdown of PRMT1 leads to substantial changes in protein arginine "methylome".
3. Discrimination of ADMA (asymmetric dimethyl arginine) and SDMA (symmetric dimethyl arginine) using characteristic neutral losses.
4. Identification of PRMT1 targets and substrate scavenged by other PRMTs in the absence of PRMT1 activity.
Protein methylation has been implicated in many important biological contexts including signaling, metabolism, and transcriptional control. Despite the importance of this post-translational modification, the global analysis of protein methylation by mass spectrometry-based proteomics has not been extensively studied because of the lack of robust, well-characterized techniques for methyl peptide enrichment.
Immunoaffinity purification (IAP), high pH strong cation exchange (SCX), and label-free quantitative proteomics.
Compared two methods for methyl peptide enrichment: IAP and SCX. In total, we identified 1720 methylation sites on 778 proteins. Notably, it revealed that these methods are largely orthogonal and quantitatively reproducible, suggesting that both methods are required for global analysis of protein methylation. PRMT1 (protein arginine methyltransferase 1) knockdown resulted in significant changes to 127 arginine methylation sites on 78 proteins. In contrast, only a single lysine methylation site was significantly changed. In PRMT1 knockdown cells, 114 monomethyl arginine sites that were either significantly down- or up-regulated on proteins enriched for mRNA metabolic processes.
Through integrative analysis of MMA and DMA, we identified a list of 18 PRMT1 substrates and 12 substrates scavenged by other PRMTs in the absence of PRMT1 activity. Taken together, our results describe a general method for deep profiling of protein methylation and identify novel potential MMA and ADMA methylation targets of PRMT1.
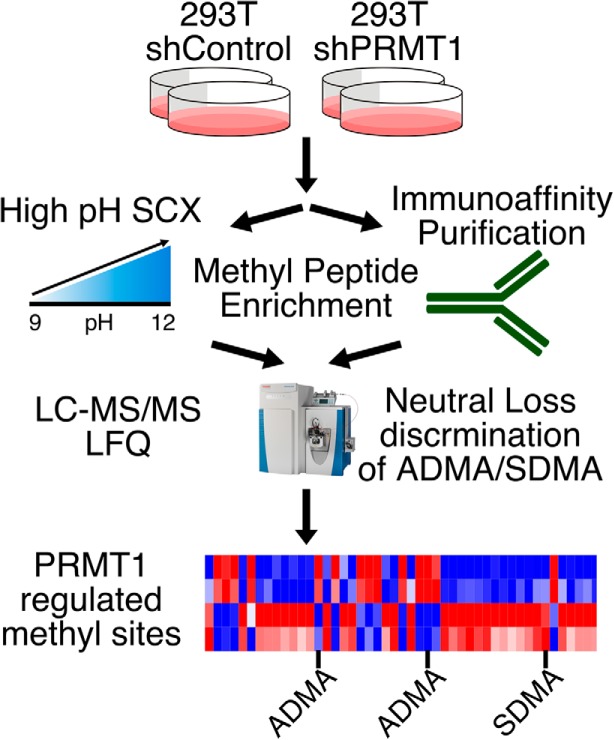 Figure 1. Workflow of quantitative proteomics of protein methylation modification.
Figure 1. Workflow of quantitative proteomics of protein methylation modification.
Publications
Here are some publications in proteomics research from our clients:

- BRCA1 antibodies matter. 2021. https://doi.org/10.7150/ijbs.63115
- Control of ribosomal protein synthesis by the Microprocessor complex. 2021. https://doi.org/10.1126/scisignal.abd2639
- Parkinson’s disease-associated LRRK2-G2019S mutant acts through regulation of SERCA activity to control ER stress in astrocytes. 2019. https://doi.org/10.1016/j.yjmcc.2007.03.738
- Confirmation of translatability and functionality certifies the dual endothelin1/VEGFsp receptor (DEspR) protein. 2016. https://doi.org/10.1186/s12867-016-0066-8
- Morphological and genetic screens reveal mechanisms of BiDAC-induced plasma membrane protein degradation. 2024. https://doi.org/10.21203/rs.3.rs-4438596/v1