
What Is 4D-Phosphoproteomics?
Standard protein expression profiling provides a foundational map, but it fails to capture functional biology. As a highly specialized high-confidence phosphosite localization service, 4D-phosphoproteomics isolates the phosphorylated sub-proteome, enabling translational research teams to move beyond baseline expression to map dynamic cellular logic.
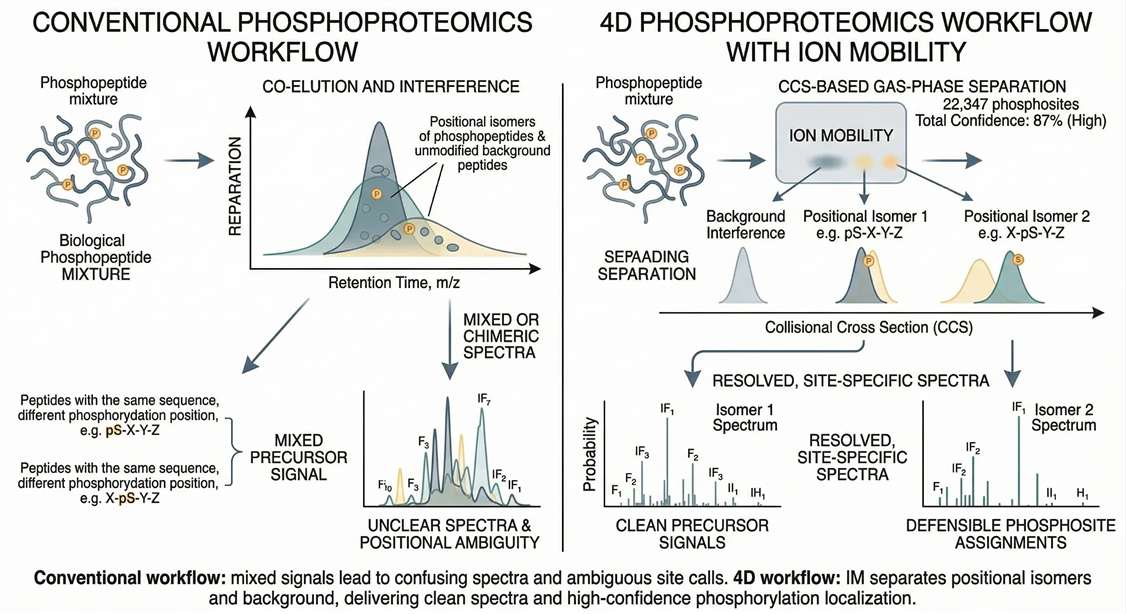
Unlike conventional workflows, this platform synchronizes LC-MS/MS with Trapped Ion Mobility Spectrometry (TIMS), introducing Collisional Cross Section (CCS) as a fourth descriptor. By filtering out co-eluting chemical noise and actively depleting unmodified background peptides through targeted enrichment, it concentrates instrument duty cycles on the low-abundance signaling events that matter.
When Total Proteomics Cannot Explain Signaling-State Biology
Rapid phenotypic transformations—such as acute therapeutic drug responses or stress adaptations—are mediated by ultra-fast signaling cascades, not slow de novo protein synthesis. Standard expression-based omics yield false-negative insights here. The mere cellular abundance of a kinase is functionally irrelevant if its critical activation loop remains unphosphorylated. Profiling these activation states reveals the "working" functional proteome.
- When abundance shifts fail: Detect low-abundance signaling events invisible to total proteomics.
- Translational mechanism research: Capture snapshots of the phosphoproteome before and after intervention.
- Drug resistance mapping: Map bypass signaling routes in resistant tumors and identify highly specific off-target kinase effects.
Content Guide
- What Is 4D-Phosphoproteomics?
- Capture Real-Time Signaling Dynamics
- What Challenges Does It Solve?
- Advantages of Our Platform
- Comparison Guide
- Step-by-Step Workflow
- Sample Requirements
- Expected Results & Deliverables
What Research Challenges Does 4D-Phosphoproteomics Solve?
From resolving positional isomers to mitigating enrichment risks, see how 4D proteomics addresses analytical roadblocks.

Positional Isomer Ambiguity
Resolves co-eluting, isobaric peptides sharing the exact amino acid sequence but carrying a phosphate group on adjacent residues (e.g., S15 vs T16) that would collapse into chimeric spectra using conventional workflows.

Isobaric Background Interference
Achieve uncompromised depth even in complex clinical matrices like FFPE or plasma, where high non-specific protein background typically limits signaling coverage or compromises site assignments.

Mitigating Enrichment-Specific Risk
For precious, low-input samples, standard workflows can cause irreversible sample loss during the enrichment phase. Our platform integrates carriers and miniaturized columns to maximize survival and defensibility.
Advantages of Our 4D-Phosphoproteomics Platform
Phosphosite Site-Level Confidence
Localization Probability > 0.75
We deploy strict algorithms to ensure reported modifications are backed by site-level FDR control below 1%.
Enrichment Specificity Controls
High-Specificity IMAC/TiO2
Targeted enrichments plus TIMS selectivity maximize specificity at low input prior to full-scale acquisition.
Low-Input Study Feasibility
Micro-Scale Protocols
Carrier strategies and micro-columns enable depth with limited clinical material.
Cohort Quantitative Stability
TIMS/RT R² ≥ 0.99
IM-aware feature alignment ensures robust reproducibility across large cohort studies.
Kinase-to-Mechanism Informatics
KSEA & Network Mapping
Integrated KSEA, motif enrichment, and pathway mapping to derive mechanism.
Discovery-to-Validation Continuity
Direct PRM Transition
Discovery outputs enable immediate targeted PRM validation assay planning.
4D-DIA vs. Standard Phosphoproteomics Comparison
| Dimension | Standard Phosphoproteomics | Discovery 4D-Phosphoproteomics | Targeted 4D-PRM Follow-Up |
|---|---|---|---|
| Site Defensibility | Low to Moderate (Prone to chimera) | High (Resolved via CCS) | Absolute (Targeted validation) |
| Positional Isomer Resolution | Weak | Excellent | Excellent (Pre-defined) |
| Input Flexibility | High inputs required (mg) | Low-input compatible (μg scalable) | Low to Moderate |
| Typical Use Case | Basic cell-line screening | Complex matrices, MOA, & tissue cohorts | Clinical biomarker panel validation |
| Mechanism-Readiness | Basic site counts | KSEA & Kinase Network Mapping | Absolute quantification of specific nodes |
| Validation-Readiness | Requires heavy manual filtering | High (Ready for PRM transition) | Final verification stage |
Selection Strategy:
- Choose 4D phosphoproteomics when site ambiguity, positional isomers, or matrix interference (e.g., in FFPE or plasma) is likely to compromise standard workflows.
- Select 4D-DIA when low-abundance signaling events are central to understanding your drug's mechanism of action.
- For total expression baselines, a standard DIA Quantitative Proteomics Service is sufficient. However, move into targeted phosphosite follow-up (such as Targeted Proteomics via 4D-PRM) when a shortlist of key regulatory nodes has been confidently defined.
Step-by-Step 4D-Phosphoproteomics Workflow
Protecting precious signaling events requires workflow control. We implement explicit study design and enrichment-aware feasibility checks.
We align on your biological goals and sample types. Tissues and cells undergo detergent-aware lysis supplemented with robust, broad-spectrum phosphatase inhibitors to instantly halt endogenous enzymatic activity and preserve the in vivo signaling state.
Because phosphopeptides represent only a tiny fraction of the total proteome, we utilize advanced IMAC (Immobilized Metal Affinity Chromatography) or TiO2 resins to selectively capture phosphorylated species while heavily washing away unmodified background peptides.
The enriched peptide pool is analyzed using TIMS-enabled mass spectrometry. Here, ion mobility provides an extra dimension of separation, cleanly resolving co-eluting positional isomers before they are fragmented, ensuring uncompromised spectral quality.
Raw data is processed via neural networks with strict <1% FDR limits. Beyond delivering filtered site matrices, we process the data through our pipeline to identify enriched motifs, infer upstream kinase activity, and map the underlying regulatory networks.
Sample Requirements and Low-Input Feasibility

Critical Handling Notes: Immediately flash-freeze all tissues and cell pellets. Ensure biological lysis buffers are heavily supplemented with robust phosphatase inhibitors.
Consultation: We offer consultation on protocol adjustments prior to shipment to maximize recovery for low-input biopsies or custom plasma designs.
| Sample Type | Typical Input | Preservation State | Main Enrichment Concern |
|---|---|---|---|
| Fresh frozen tissue | 1-2 mg | Flash-frozen immediately | Endogenous phosphatase degradation |
| FFPE | 5-15 curls (10μm) | Mounted/Unmounted | Low yield due to cross-linking |
| Cells / organoids | 106 - 107 cells | Washed cell pellet | Lysis buffer interference |
| Low-input biopsy | Consult required | Snap-frozen | Irreversible sample loss |
| Serum / plasma | 100-300 μL | Unhemolyzed, spun | Severe high-abundance interference |
Not sure whether your samples meet the requirements?
Contact us — we're happy to help design the best strategy for your 4D-Phosphoproteomics study.
Expected Results and Bioinformatics Deliverables
A list of phosphosites is just the starting point. Our integrated Proteomics Bioinformatics Analysis Service pipeline transitions broad data lists into mechanism-oriented decisions.
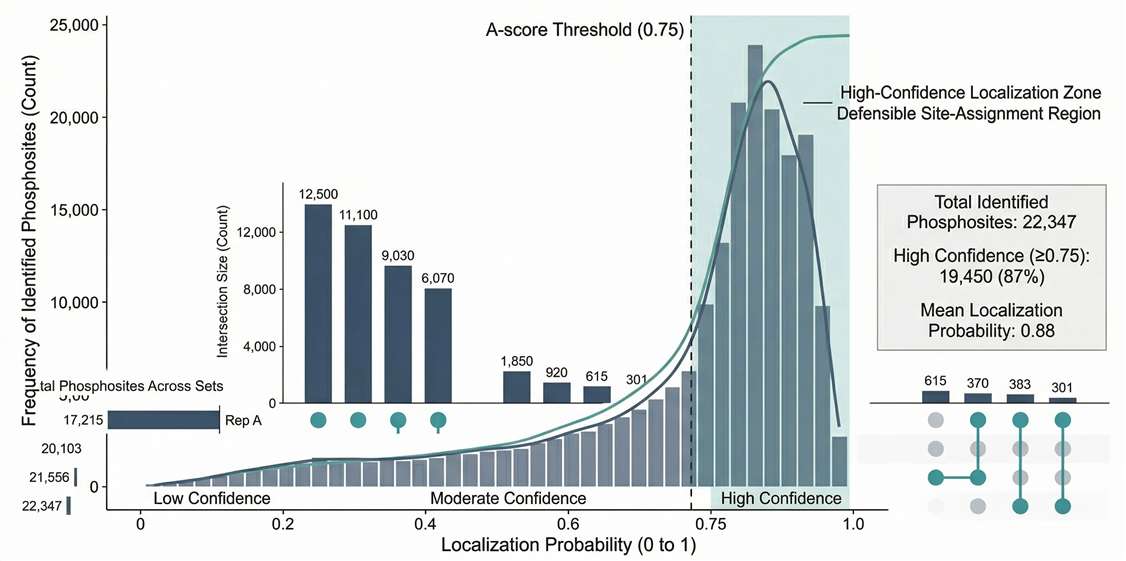
Localization Confidence Distribution: Validates dataset defensibility by showing the high proportion of sites meeting strict probability thresholds.
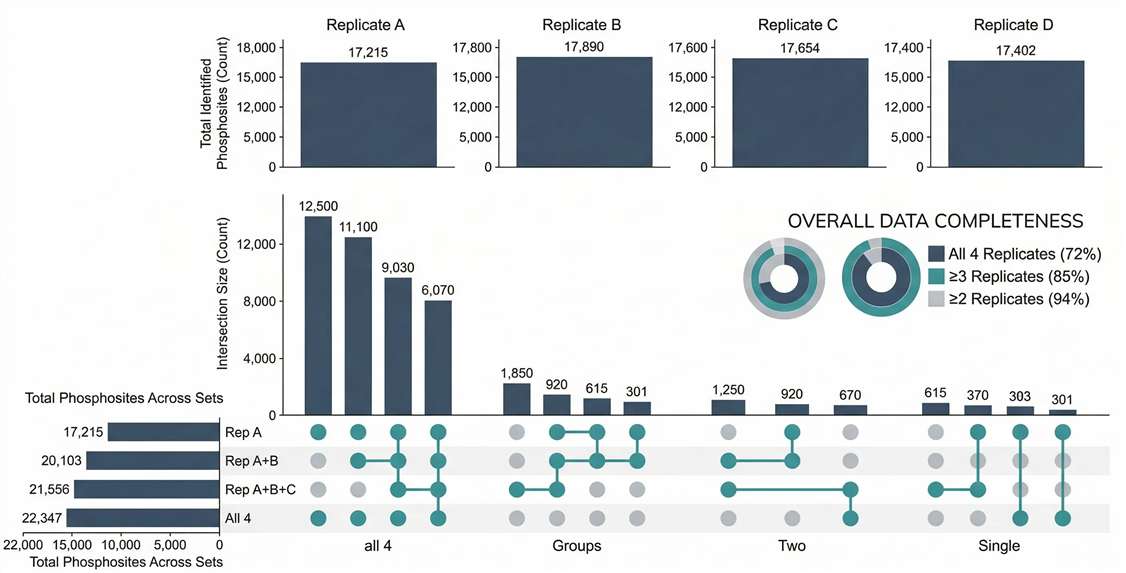
Coverage & Overlap Analysis: Confirms high enrichment specificity and quantitative overlap across biological replicates.
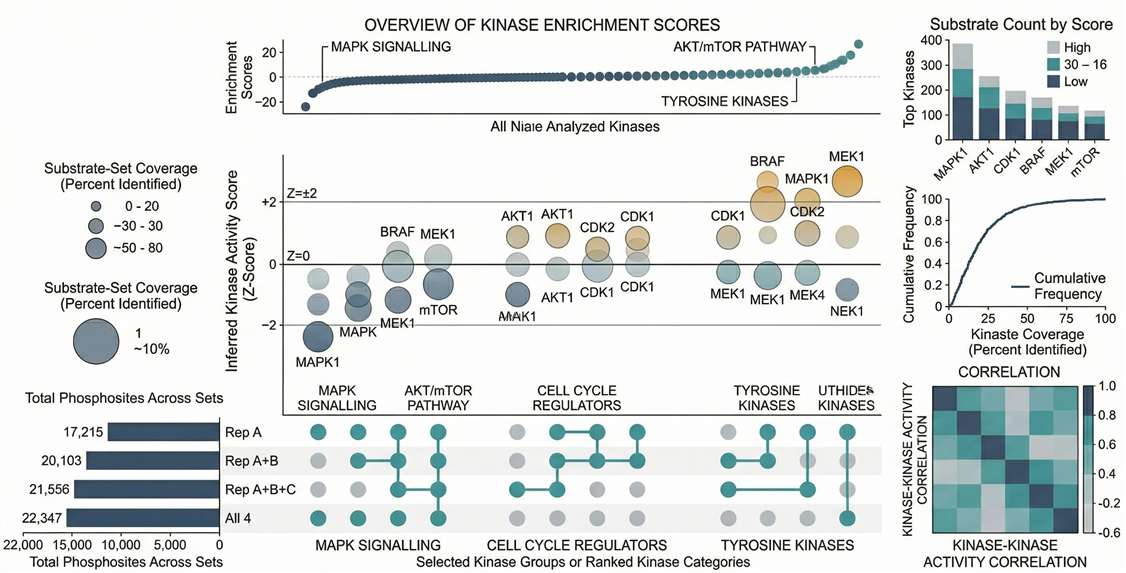
Kinase-Substrate Enrichment (KSEA): Ranks the most significantly activated or inhibited upstream kinases based on their substrate networks.
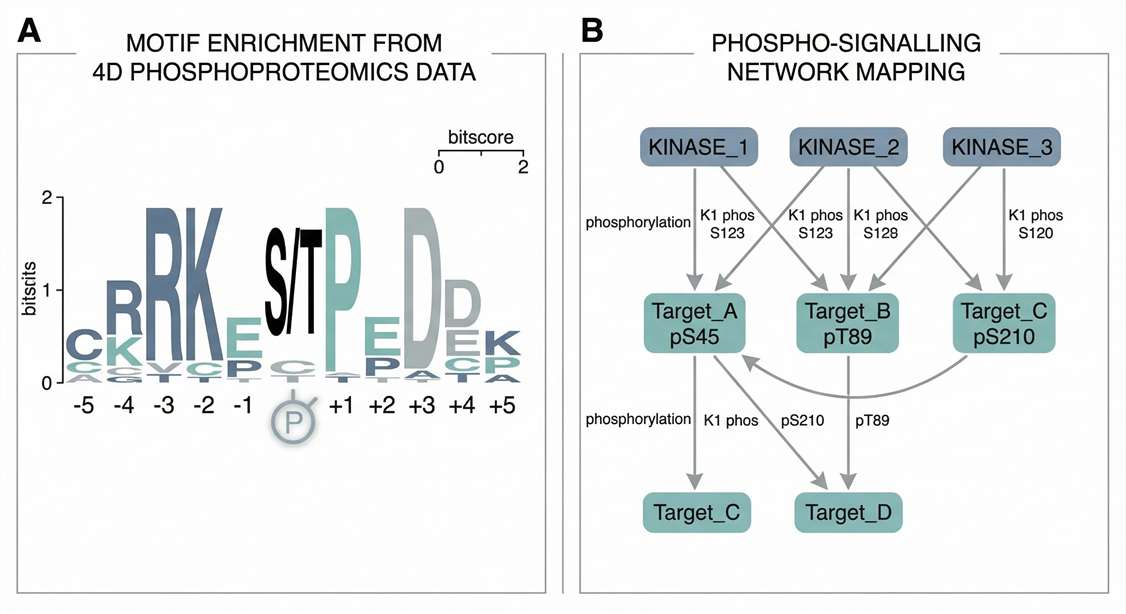
Motif & Network Mapping: Visualizes phospho-protein interactions and extracts target enzyme sequence signatures driving the cellular response.
Evidence: Deep and Reproducible 4D-Style Phosphoproteomics for Signaling Studies
A data-independent acquisition-based global phosphoproteomics system enables deep profiling
Journal: Nature Communications · Published: 2021
Study Scope
Researchers developed a global phosphoproteomics system (GPS) based on DIA-MS to achieve deep, accurate, and reproducible phosphosite profiling in cancer-relevant samples. Using non-small cell lung cancer (NSCLC) as a model, the study combined direct DIA and library-based DIA with a high-quality hybrid phosphoproteome spectral library to improve site localization, depth, and quantitative robustness.
- The study established a phosphoproteome spectral library containing 159,524 phosphopeptides on 8,805 protein groups, covering 88,107 phosphosites.
- A single-shot DIA workflow achieved deep quantification of 38,255 phosphosites, including 20,420 class 1 sites.
- The method was benchmarked with 166 synthetic phosphopeptides relevant to lung cancer signaling to assess analytical performance.
- The workflow incorporated consistent chromatography, iRT peptides, Fe-IMAC phosphopeptide enrichment, and hybrid spectral-library construction to improve data quality and reproducibility.
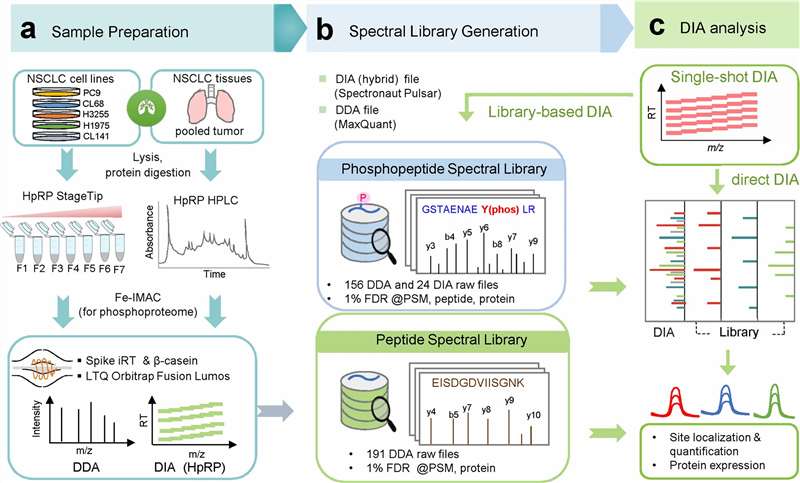 Workflow of the DIA-based global phosphoproteomics strategy used for deep and reproducible phosphosite profiling.
Workflow of the DIA-based global phosphoproteomics strategy used for deep and reproducible phosphosite profiling.
Phosphosite Confidence and Quantitative Performance
The study directly addressed several of the concerns that matter most in phosphoproteomics projects: site-level confidence, reproducibility, overlap, and missing-value reduction. The authors reported that 161 of 166 phosphosites (96%) showed high accuracy for class 1 site determination at localization probability ≥ 0.75, with strong agreement between measured and library-annotated retention times (R² = 0.996).
Additional performance comparisons showed that library-based DIA substantially improved phosphosite depth and reproducibility relative to DDA and direct DIA:
- Phosphosite overlap between direct DIA and library-based DIA was explicitly compared.
- Median phosphosite CV values were reported as 13.0% for DDA, 4.3% for direct DIA, and 5.2% for library-based DIA, highlighting much stronger quantitative consistency for DIA-based phosphoproteomics.
- The authors also reported lower between-run missing values and strong sensitivity, especially for phosphotyrosine-relevant coverage.
These results are highly relevant for advanced phosphoproteomics decision-making because they show that deeper phosphosite coverage is only valuable when it is paired with strong localization accuracy, overlap between runs, and quantitative reproducibility.
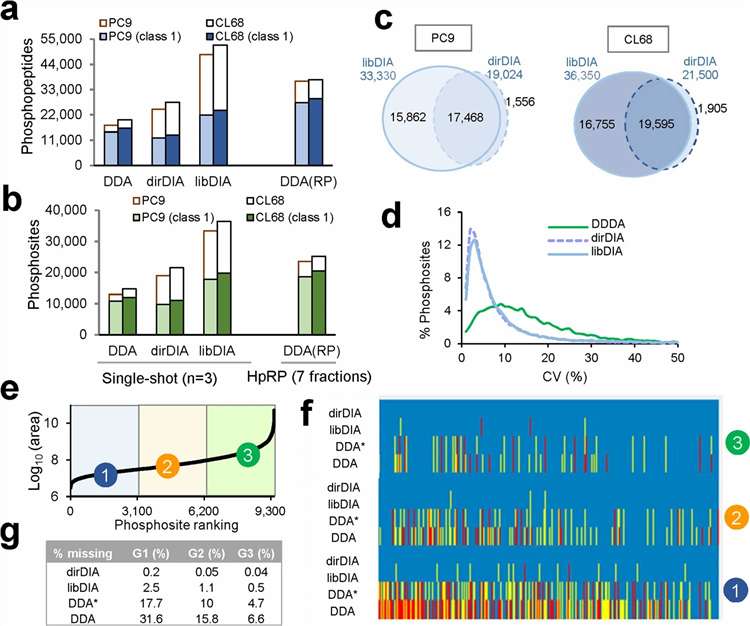 DIA-based phosphoproteomics improves phosphosite depth, overlap, and quantitative reproducibility relative to conventional workflows.
DIA-based phosphoproteomics improves phosphosite depth, overlap, and quantitative reproducibility relative to conventional workflows.
Why This Case Matters for 4D-Phosphoproteomics
This case is a strong fit for a phosphoproteomics service page because it demonstrates the exact performance dimensions buyers care about most:
- Phosphosite defensibility, not just site counts
- Localization-confidence control for downstream validation
- Lower missing values and stronger quantitative reproducibility across runs
- Deep pathway-relevant phosphoproteome coverage in disease-focused samples
Together, these results support the value proposition of advanced phosphoproteomics workflows for signaling studies, kinase-pathway analysis, and discovery programs that need a credible bridge to targeted follow-up.
Reference
Gao, Y. et al. "A data-independent acquisition-based global phosphoproteomics system enables deep profiling." Nature Communications 12, 2539 (2021).