
Principles of SWATH-MS Label-Free Quantification
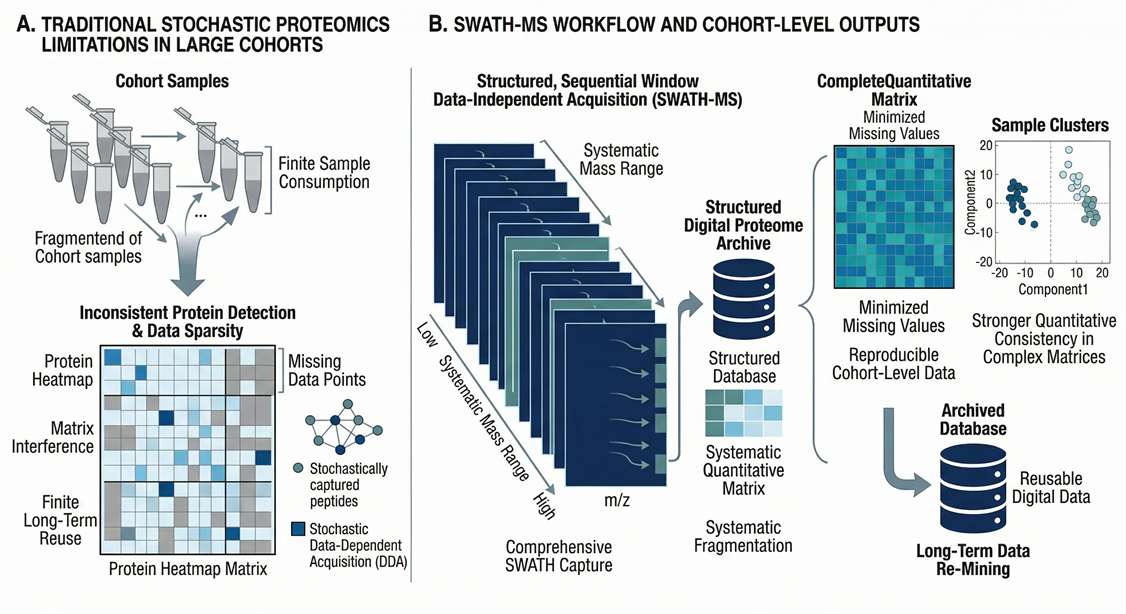
SWATH-MS (Sequential Window Acquisition of all Theoretical Mass Spectra) is a premier form of Data-Independent Acquisition (DIA). Unlike traditional Data-Dependent Acquisition (DDA)—which stochastically fragments only the most intense precursor ions and often misses low-abundance proteins—SWATH-MS systematically fragments all ionizable peptides within predefined isolation windows (e.g., 20–25 Da) across the entire mass range.
By fragmenting every co-eluting peptide simultaneously, it produces a continuous, unbiased digital map of the proteome. We then utilize high-quality, project-specific spectral libraries to decode these multiplexed MS2 spectra. This "targeted data extraction" approach transforms complex raw data into precise quantitative profiles, beautifully combining the broad scale of global discovery proteomics with the analytical rigor of targeted methods.
Content Guide
- Principles of SWATH-MS
- SWATH-MS vs. DDA
- Retrospective Data Mining
- Research Challenges Solved
- Key Applications
- Service Advantages
- Technology Selection Guide
- Performance Specifications
- Assay Development Workflow
- Sample Requirements
- Bioinformatics & Deliverables
SWATH-MS vs. Traditional DDA: Solving the "Missing Value" Crisis
The "missing value" crisis often causes large-scale cohort studies to fail during biomarker validation. Standard DDA relies on random intensity sampling, meaning a biomarker sequenced in one sample might be completely missed in another due to matrix interference. Scaling to hundreds of samples amplifies these data gaps, destroying statistical power.
SWATH-MS solves this by recording all fragment ions within sequential isolation windows regardless of precursor intensity. If a peptide is physically present, it is permanently recorded and quantified. This delivers near 100% data completeness across massive cohorts, ensuring robust statistical power for clinical biomarker ranking and longitudinal studies.
| Feature | SWATH-MS (DIA) | Traditional DDA | Targeted MS (PRM/MRM) |
|---|---|---|---|
| Data Completeness | Near 100% (No missing values) | Low (Stochastic dropouts) | 100% (For predefined targets only) |
| Acquisition Strategy | Unbiased (Fragments all ions) | Biased (Selects intense peaks) | Highly Biased (Targets only) |
| Multiplexing Limit | Unlimited (Global profiling) | Unlimited (Global profiling) | Limited (~50-150 proteins) |
| Retrospective Mining | Yes (Digital Archive) | No (Unfragmented data lost) | No (Only targets recorded) |
| Inter-sample CV | Excellent (Consistent < 10%) | Poor-to-Moderate | Excellent (CV < 10%) |
Retrospective Data Mining in SWATH-MS Proteomics
Our SWATH-DIA service creates a Permanent Digital Archive of your research cohorts. For finite clinical materials like needle biopsies, cerebrospinal fluid (CSF), sorted cell populations, or highly specific longitudinal plasma samples, traditional ELISAs and targeted assays consume the material entirely, leaving nothing for future verification.
- Archived Raw Data: Every SWATH-MS run records a complete physical snapshot of the entire proteome at that exact moment in time, saved securely as a permanent `.wiff` file.
- In-Silico Re-Extraction: If a new biological signaling pathway is published in the literature three years after your project concludes, you do not need to re-run physical samples.
- Target Flexibility: We simply update the spectral reference library with the new protein targets and extract the quantitative data directly from the archived files.
This "measure once, mine forever" strategy ensures that your study remains biologically relevant even as new scientific breakthroughs occur, providing an unparalleled return on investment for your clinical cohorts.
What Research Challenges Does Our SWATH-MS Service Solve?

Statistical Power Failure
Eliminates the missing value problem that causes large-scale clinical validation to fail due to sparse and incomplete data matrices.

Finite Sample Depletion
Digitizes rare or precious clinical samples in a single injection, allowing for limitless future querying without consuming additional physical material.

TMT Batch Effect Limitations
Avoids the ratio compression artifacts and inter-kit batch effects of isobaric labeling (TMT), making it the gold standard for label-free studies exceeding 50 samples.
High Complexity Matrix Noise
Utilizing high-resolution isolation windows to resolve low-abundance tissue-leakage proteins that are often obscured in deep plasma or un-depleted agricultural samples.
Key Applications of SWATH-MS Proteomics
Clinical Biomarker Discovery
Consistent identification and validation of candidate biomarker panels across massive independent cohorts of plasma, serum, or FFPE tissues, maintaining statistical rigor across hundreds of patients.
AgBio & Plant Stress Response
Highly reproducible profiling of crop stress responses, breeding traits, and secondary metabolic pathways across diverse agricultural populations, overcoming extreme dynamic range challenges in plant tissues.
PK/PD & Global Toxicology
Longitudinal monitoring of drug-induced protein expression shifts and global assessment of off-target toxicity in pharmaceutical development without being restricted to pre-defined target panels.
Microbiome & Metaproteomics
Quantifying complex host-pathogen interactions and deciphering intricate microbial community dynamics with robust quantitative consistency across highly heterogeneous multi-species environments.
Advantages of Our SWATH-DIA Biomarker Discovery Service
Deep Custom Libraries
We perform offline 2D-LC fractionation on pooled cohort samples to build project-specific reference libraries, maximizing proteome extraction depth.
Zero Missing Values
Capture every fragment ion systematically to deliver gap-free quantitative matrices, rescuing low-abundance biomarkers missed by top-down DDA.
Permanent Digital Archive
Establish an everlasting digital biobank of your proteome, supporting retrospective mining of new targets as your research focus evolves.
Cohort Reproducibility
Extreme technical consistency via SCIEX platform optimization and iRT standard spike-ins, maintaining technical CVs below 10%.
Advanced Bioinformatics
Comprehensive deliverables including PCA, KEGG enrichment, volcano plots, and ROC curve evaluation for biomarker clinical utility.
Infinite Scalability
Our label-free workflow bypasses TMT batch effect limitations, making it the premier choice for large-scale studies exceeding 100 samples.
Technology Selection Guide: When to Choose SWATH-MS
Method selection should be driven by panel size, matrix complexity, quantitative objective, and whether the project is in candidate verification, cohort expansion, or concentration-level reporting.
SWATH-MS vs. DDA vs. General DIA
| Dimension | SWATH-MS (Library-Based DIA) | Traditional DDA | General Library-Free DIA |
|---|---|---|---|
| Data Completeness | Near 100% (No missing values in large cohorts) | Prone to stochastic dropouts | High, but sensitive to false discoveries |
| Quantification Approach | Targeted extraction via empirical spectral libraries | Extracted ion chromatograms (XIC) of MS1 | Direct spectrum prediction (In silico) |
| Best Used For | Large-cohort label-free quant with strict consistency | Standard deep profiling (<20 samples) | Rapid discovery without pooled sample prep |
| Library Requirement | Project-specific 2D-LC library recommended | Not required | None (Algorithm-based prediction) |
SWATH-MS vs. Targeted PRM/MRM vs. 4D-DIA
| Dimension | SWATH-MS | Targeted PRM/MRM | 4D-DIA (PASEF) |
|---|---|---|---|
| Primary Objective | Broad, unbiased global discovery | Absolute quantitation of defined targets | Maximum depth in complex matrices |
| Multiplexing Limit | Unlimited | ~50–150 targets per run | Unlimited |
| Key Technological Advantage | Cohort-level consistency & digital archiving | Absolute molar quantification (fmol/μL) | Ion Mobility (CCS) isomer separation |
| Ideal Project Phase | Biomarker discovery & cohort expansion | Late-stage clinical validation | Deep discovery in un-depleted plasma |
- Choose SWATH-MS when you have a massive clinical cohort (n > 50), require a permanent digital archive for future re-analysis, and want to completely eliminate the missing value problems inherent to DDA.
- Choose Targeted PRM/MRM when you have already identified a specific panel of 10 to 100 targets and require absolute molar quantification (e.g., fmol/μL) using heavy-isotope AQUA internal standards for validation.
- Choose 4D-DIA (PASEF) when you are dealing with severely complex matrices (like un-depleted plasma) and need Ion Mobility (CCS values) to physically separate co-eluting isomeric peptides in the gas phase.
Assay Performance & Validation Specifications
We hold our SWATH-MS analytical workflows to stringent quality control standards, ensuring your high-throughput data is unassailable and ready for publication:
- Deepest Project-Specific Coverage: Offline 2D-LC fractionated DDA library generation tailored to your exact cohort significantly outperforms generic pan-human libraries by accounting for sample-specific splice variants and background matrix.
- Cohort-Grade Consistency: Implementation of indexed Retention Time (iRT) standards ensures precise chromatogram alignment, maintaining median CVs < 15% across massive multi-month cohort acquisitions.
- Rigorous FDR Control: Advanced statistical algorithms (such as mProphet) are employed for peak scoring and validation, ensuring a false discovery rate (FDR) strictly below 1% at both the peptide and protein levels.
- Expert Support: End-to-end scientific guidance from initial power analysis and cohort study design through to deep biological pathway interpretation.
End-to-End SWATH-DIA Biomarker Discovery Workflow
Our facility executes a rigorously controlled process to transform raw biological material into a comprehensive digital proteome map.
Optimized lysis, reduction, alkylation, and digestion protocols for your specific matrix. QC: Assessment of peptide yield, purity, and missed-cleavage rates to guarantee digestion efficiency.
Offline high-pH reverse-phase fractionation of representative cohort pools is subjected to deep DDA analysis to build a highly specific, high-resolution spectral reference library.
Individual samples are acquired using variable isolation window widths optimized for peak precursor density on high-resolution platforms. QC: Continuous monitoring of spiked iRT peptides to verify LC stability.
Complex multiplexed spectra are decoded via the custom library. Data undergoes robust FDR filtering, normalization, and sophisticated statistical modeling to isolate true biological variance.
Sample Requirements for SWATH-MS Analysis
SWATH-MS is highly versatile and supports virtually any biological matrix. For biofluids, we typically recommend high-abundance protein depletion to maximize the detection depth of the low-abundance tissue-leakage proteome.
| Sample Type | Minimum Amount | Recommended Amount | Preservation State |
|---|---|---|---|
| Plasma / Serum | 20 μL | 50 - 100 μL | Flash-frozen, Unhemolyzed |
| Fresh Frozen Tissue | 5 mg | 10 - 20 mg | Flash-frozen (Liquid N2) |
| FFPE Tissue | 3-5 curls (10μm) | 5-10 curls (10μm) | Mounted or Unmounted |
| Plant Samples | 50 mg | 100 - 200 mg | Flash-frozen |
| Cell Lines | 106 cells | 5 x 106 cells | Washed cell pellet, frozen |
SWATH-MS Bioinformatics Analysis & Data Deliverables
Our bioinformatics analysis and reporting workflow focuses on analytical performance, quantitative clarity, and validation-ready interpretation. We deliver the statistical evidence you need to confidently advance your translational pipeline.
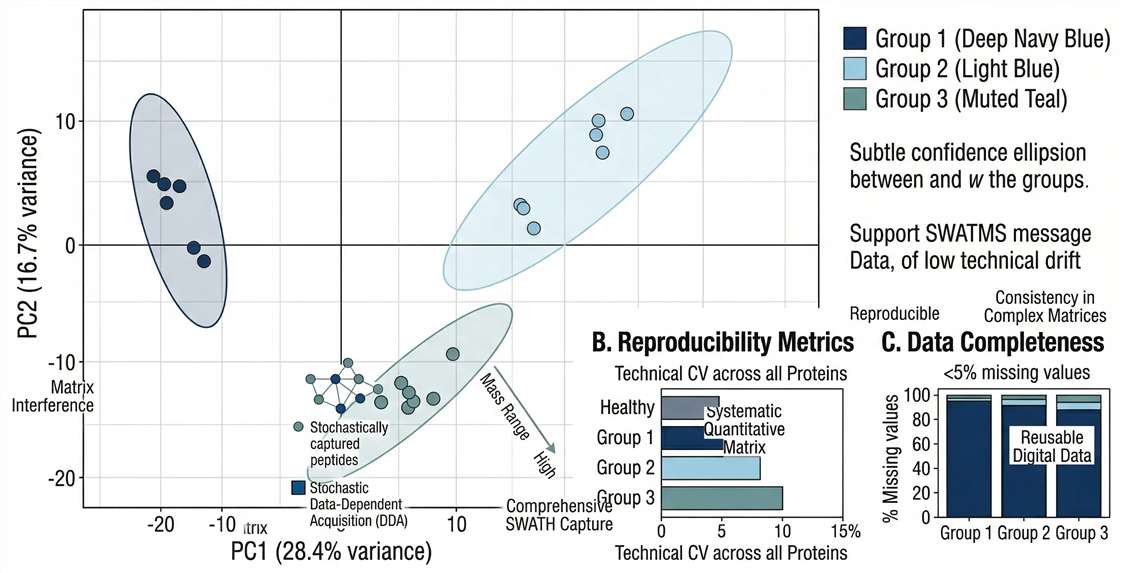
Data Quality & Reproducibility: Principal Component Analysis (PCA) plots demonstrating tight clustering of biological replicates and effective batch effect elimination.
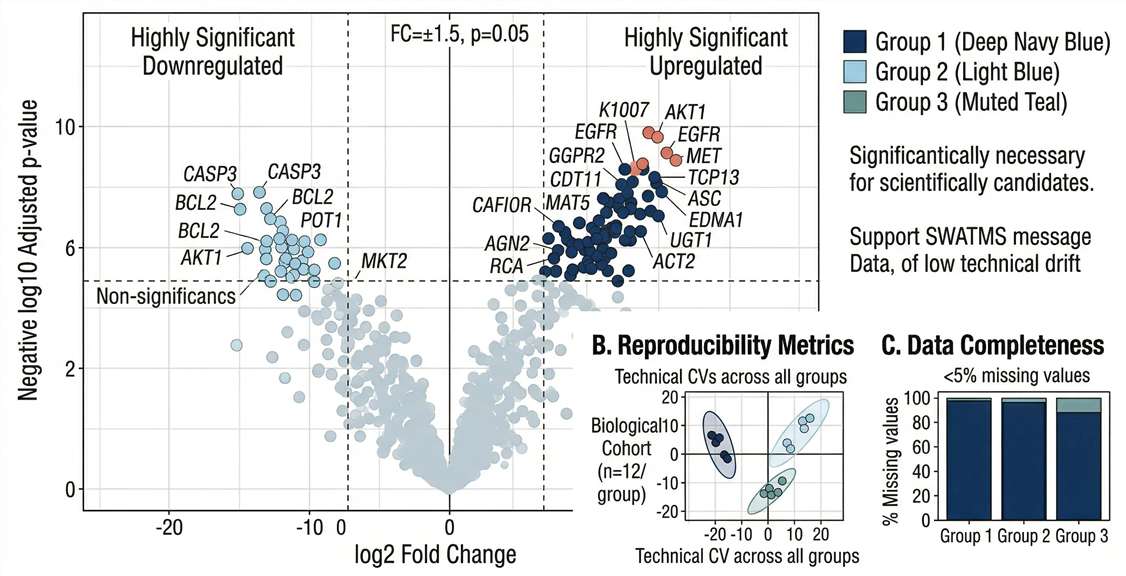
Differential Expression: High-resolution Volcano plots clearly delineating statistically significant biomarker candidates based on fold-change and p-value.
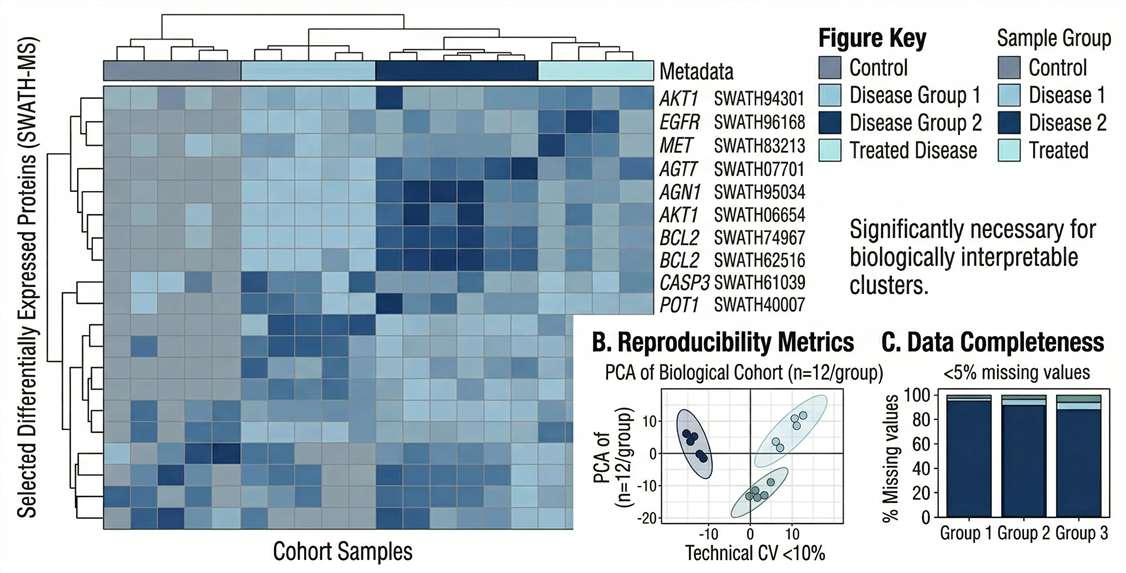
Expression Patterns: Hierarchical clustering heatmaps illustrating distinct patient stratification between diseased groups and healthy controls.
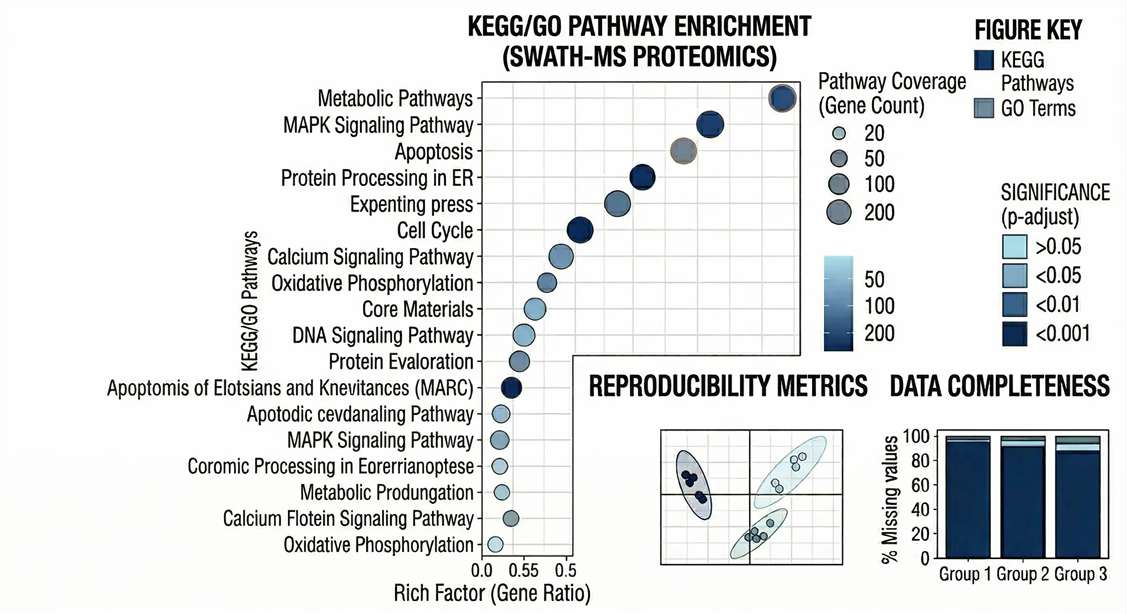
Functional Enrichment: KEGG and GO pathway enrichment bubble charts revealing the core biological drivers and signaling networks.
Raw Data Files (.wiff)
Native SWATH files serving as your permanent digital archive for future retrospective mining.
Processed Quantitative Tables
Clean, missing-value-filtered matrices containing normalized protein intensities for all cohort samples.
Quality Control Pack
Detailed technical reports on peptide/protein ID rates, FDR thresholds, and iRT alignment metrics.
Technical Reports
Ready-to-publish methodology sections, high-resolution figures, and deep biological pathway interpretations.
Representative Clinical Biomarker Discovery Using SWATH-MS
Blood-based protein biomarkers for the diagnosis of acute stroke: A discovery-based SWATH-MS proteomic approach
Journal: Frontiers in Neurology · Published: 2022
Study Scope
In this discovery-phase clinical study, researchers used SWATH-MS to identify blood-based protein biomarker candidates for acute stroke diagnosis within 24 hours of symptom onset. Serum samples from ischemic stroke (IS), intracerebral hemorrhage (ICH), and matched healthy controls were analyzed without pooling, allowing subject-level quantitative comparison rather than averaged group-level signals.
- The study included 40 stroke cases (20 IS and 20 ICH) and 40 matched healthy controls.
- A high-pH fractionated human serum peptide library was used to support SWATH-MS-based quantitative extraction.
- The workflow was designed to discover differentially expressed proteins and nominate candidate diagnostic biomarkers in an acute clinical setting.
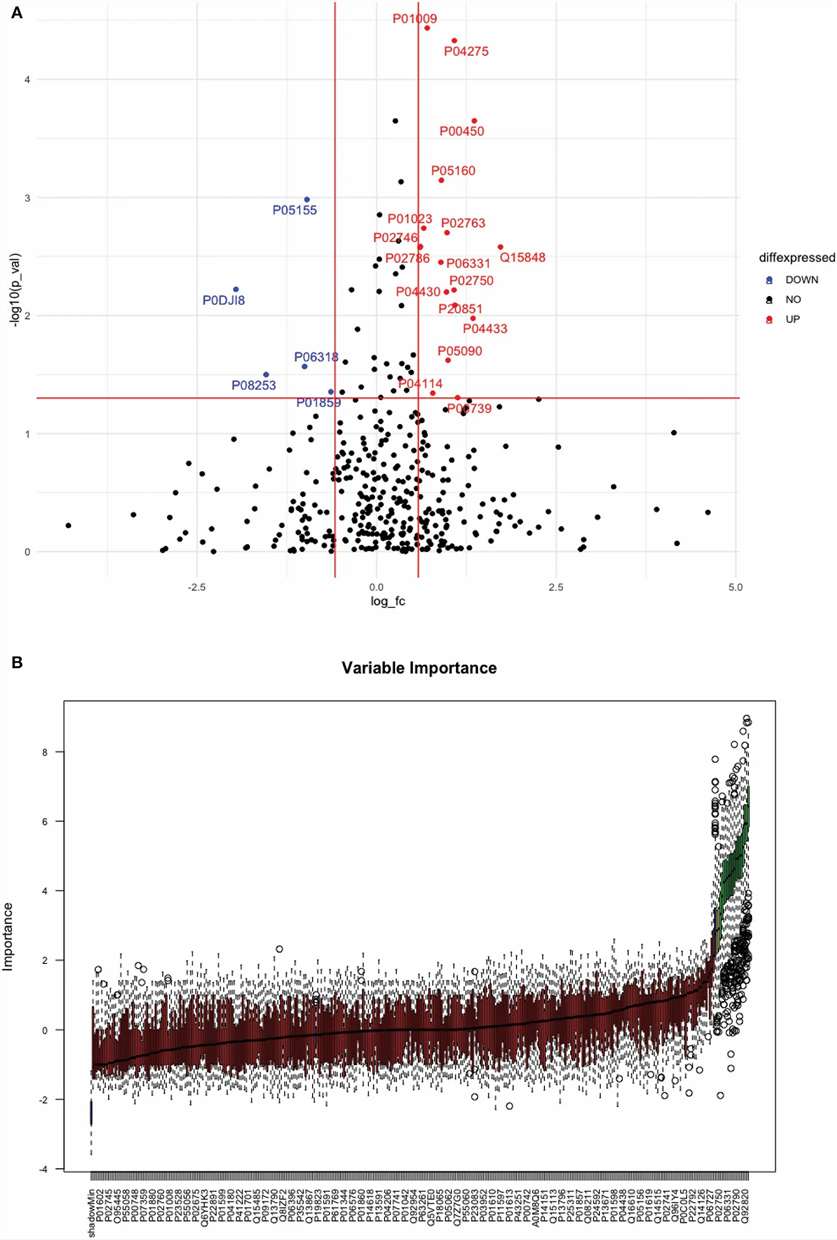 Figure 2. Volcano plot and Boruta random forest feature selection identify significantly differentially expressed proteins between total stroke cases and healthy controls.
Figure 2. Volcano plot and Boruta random forest feature selection identify significantly differentially expressed proteins between total stroke cases and healthy controls.
Quantitative Performance and Biological Findings
Using SWATH-MS, the researchers quantified 375 proteins across the serum cohort. They identified 31 significantly differentially expressed proteins between total stroke and healthy controls, 16 proteins between ischemic stroke and controls, and 41 proteins between intracerebral hemorrhage and controls.
- Four proteins—ceruloplasmin, SERPINA1, von Willebrand factor (vWF), and factor XIII B chain (F13B)—commonly differentiated total stroke, IS, and ICH from healthy controls.
- Heatmap analysis showed that the significantly differentially expressed proteins could separate stroke samples from control samples at the cohort level.
- Protein-protein interaction and enrichment analysis highlighted biologically coherent pathways such as complement and coagulation cascades, platelet degranulation, and acute-phase / immune-related processes.
This study is a strong example of how SWATH-MS supports discovery-stage biomarker prioritization in clinical cohorts while preserving subject-level quantitative consistency.
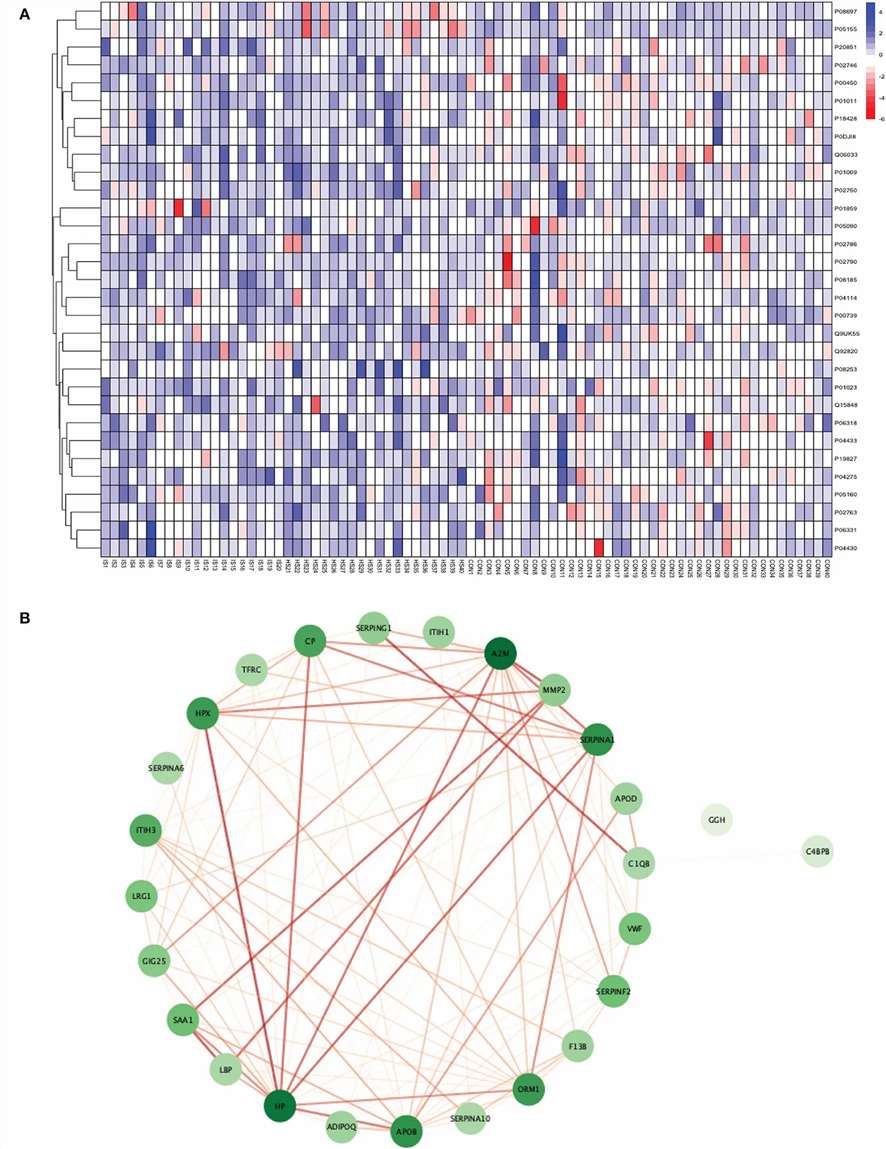 Figure 3. Heatmap and protein-protein interaction network analysis show cohort-level separation and biologically connected protein changes in total stroke versus healthy controls.
Figure 3. Heatmap and protein-protein interaction network analysis show cohort-level separation and biologically connected protein changes in total stroke versus healthy controls.
Why This Case Matters for Your SWATH-MS Strategy
This paper illustrates the real value of SWATH-MS for translational biomarker discovery: reproducible cohort-level quantification, broad protein coverage from serum samples, and downstream biological interpretation without relying on stochastic DDA sampling. For teams planning medium-to-large discovery cohorts, it demonstrates how SWATH-MS can generate statistically tractable candidate biomarker lists while preserving the opportunity for later targeted validation.
- Supports cohort-scale label-free discovery with subject-level quantitative consistency
- Enables biomarker candidate prioritization from complex serum samples
- Provides pathway-level biological interpretation beyond simple protein lists
- Creates a discovery dataset that can later inform targeted PRM/MRM follow-up
Reference
Misra, S., Singh, P., Nath, M., Bhalla, D., Sengupta, S., Kumar, A., Pandit, A. K., Aggarwal, P., Srivastava, A. K., Mohania, D., Prasad, K., and Vibha, D. "Blood-based protein biomarkers for the diagnosis of acute stroke: A discovery-based SWATH-MS proteomic approach." Frontiers in Neurology 13 (2022): 989856.