
What Is 4D PTM Proteomics?
How 4D PTM proteomics adds a regulatory layer beyond total proteome profiling
While total proteomics quantifies baseline protein expression, it cannot reveal the activation state or spatial organization of those proteins. A post-translational modification proteomics service captures the dynamic regulatory layer of the proteome. By integrating Trapped Ion Mobility Spectrometry (TIMS) into LC-MS/MS workflows, 4D PTM proteomics isolates and quantifies the exact modified peptides driving cellular function.
This 4D separation delivers critical advantages for mechanism-focused research:
- CCS-Supported Matching: Adds Collisional Cross Section (CCS) as a fourth dimension to physically separate isobaric positional isomers before fragmentation, radically improving enrichment selectivity.
- Site-Level Confidence: Ensures uncompromising precision when identifying the exact amino acid residue modified, eliminating ambiguity in downstream mutagenesis.
- Regulatory Profiling: Uncovers dynamic molecular switches governing signaling cascades, targeted proteasomal degradation, and chromatin structural states.
Content Guide
- What Is 4D PTM Proteomics?
- Why PTM Analysis Matters
- Challenges 4D PTM Can Solve
- Choose the Right PTM Route
- Why 4D PTM is a Better Choice
- Advantages of Our Platform
- Platforms & Enrichment Strategies
- Discovery vs Targeted Guide
- Step-by-Step Workflow
- Sample Requirements
- Expected Results & Deliverables
Why PTM Analysis Matters When Total Proteomics Is Not Enough
Researchers frequently observe dramatic phenotypic changes—such as acute therapeutic drug responses, stress adaptations, or rapid disease progression—while global total protein levels remain static. This occurs because the initial biological response is mediated by rapid, transient post-translational events, not by de novo protein synthesis. In these scenarios, a standard expression-based omics approach will yield false-negative insights.
Enzyme activation, protein-protein interaction affinities, and subcellular localization are strictly dictated by modification states. Profiling the modification state reveals the actual "working" functional proteome. A dedicated 4D proteomics platform allows researchers to selectively enrich specific regulatory sub-proteomes, providing the high-resolution data required to deconvolve complex signaling networks, investigate proteostasis, and decipher epigenetic regulation.
What Research Challenges Can 4D PTM Proteomics Help Solve?

Low-Abundance Signals Masked by Background
Modifications are inherently substoichiometric. In complex matrices like plasma or tissue lysates, crucial regulatory signals are drowned out. 4D workflows utilize ion mobility to separate chemical noise, effectively pushing critical low-abundance signals above the interference threshold.

Poor Specificity & Weak Site Confidence
Noisy site tables filled with false-positive assignments inevitably ruin downstream validation experiments. We utilize 4D TIMS separation to provide a definitive physical validation (CCS) of the modified peptide, ensuring stable and defensible site calls.

Low-Input Limits with Precious Samples
Traditional enrichment chemistry demands milligram-level protein inputs. Our micro-scale PTM strategies are designed to aggressively mitigate sample loss during lysis and enrichment, answering the question: "Will my sample survive?"

Interpretation Gaps (Tables vs. Biology)
Delivering an unfiltered spreadsheet of modified sites leaves researchers with a severe interpretation gap. We bridge this by transitioning raw numeric matrices into route-specific biological models (e.g., precise kinase-substrate mapping).
How to Choose the Right PTM Route for Your Study
Selecting the right PTM route is often the most important decision at the start of a regulatory proteomics project. Different modification classes answer different biological questions, require different enrichment logic, and support different downstream interpretation paths. The guide below helps map your research objective to the most appropriate PTM workflow and downstream service page.
| Research Focus | Key Biological Mechanism | Best PTM Route |
|---|---|---|
| Signal transduction, pathway activation, and kinase response | Receptor signaling, kinase-substrate activity, rapid regulatory switching | 4D-Phosphoproteomics |
| Protein degradation, proteostasis, and targeted degrader studies | E3 ligase biology, ubiquitin remnant mapping, PROTAC-related regulation | 4D-Ubiquitination Proteomics |
| Chromatin regulation, transcriptional control, and metabolic rewiring | Histone acetylation, acetyl-CoA–linked regulation, enzyme-state modulation | 4D-Acetylation Proteomics |
| Regulatory protein modification and methylation-driven control | Lysine/arginine methylation, transcriptional regulation, protein-state control | 4D-Methylation Proteomics |
These four routes represent the most common entry points for signaling, degradation, chromatin, and regulatory PTM studies. Additional or combined PTM designs can also be evaluated based on biological question, sample type, enrichment feasibility, and downstream validation goals.
Why 4D PTM Proteomics Is a Better Choice Than Conventional PTM Workflows
For PTM studies, the difference is not just identification depth. The critical question is whether modified peptide assignments remain defensible in the presence of isobaric interference, low-abundance signals, and difficult sample backgrounds. 4D PTM workflows add a physical separation dimension that improves site-level confidence and supports more stable downstream interpretation.
What Changes in 4D PTM
Ion mobility adds the Collisional Cross Section (CCS) as an orthogonal physical descriptor. This critical dimension helps cleanly separate co-eluting modified peptides prior to fragmentation, significantly reducing ambiguity in site assignment.
Why It Matters
This is especially valuable when target modified peptides are extremely low in abundance, when the enrichment background remains high, or when multiple positional isomers would otherwise collapse into less-defensible mass spectra.
When 4D Is Worth It
Choose 4D PTM workflows when absolute site defensibility, robust low-input sample compatibility, and mechanism-ready biological interpretation matter far more than merely generating a larger, yet ultimately less stable and unverified modification list.
Advantages of Our High-Confidence 4D PTM Proteomics Platform
Localization-Filtered Site Reporting
Strict Probability Scoring
We enforce strict localization probability scoring algorithms and physical CCS validation to ensure only defensible, high-confidence sites reach your final biological report.
Enrichment-Specific QC Checkpoints
Pre-Acquisition Verification
We rigorously verify the specificity of your enriched peptide pool prior to full-scale MS acquisition, mitigating the risk of analyzing noisy, unmodified background data.
Low-Input-Compatible Workflows
Micro-Scale Adaptation
Subject to our mandatory sample-type feasibility review, our optimized micro-scale enrichment protocols adapt successfully to precious clinical biopsies and sorted cell populations.
Cohort-Aware PTM Quantitation
IM-Aware Feature Alignment
We utilize sophisticated IM-aware alignment to manage missing values intelligently across analytical batches, ensuring longitudinal consistency for large translational cohorts.
Mechanism-Ready Analysis
Biological Pathway Inference
We expertly transition your data from raw site matrices into actionable biological pathways, algorithmically inferring upstream regulators that drive the observed phenotype.
Child-Page Continuity
Seamless Subtype Routing
Our parent hub acts as a dedicated consultative guide, helping you confidently evaluate whether phosphorylation, ubiquitination, or another PTM best answers your hypothesis.
Advanced Platforms and Enrichment Strategies
A successful PTM project requires the perfect marriage of front-end biochemical enrichment chemistry and back-end mass spectrometry acquisition power. Our advanced services are anchored by the timsTOF Pro 2 (Bruker) for true 4D-DIA (diaPASEF) capabilities, complemented by the Orbitrap Exploris 480 (Thermo) for standard baseline DIA and highly targeted PRM validation panels.
Because different modifications exhibit unique stoichiometric challenges and biochemical properties, we pair our mass spectrometry platforms with highly specific enrichment logic by PTM class:
| PTM Class | Primary Enrichment Chemistry | How 4D (Ion Mobility) Enhances Results |
|---|---|---|
| Phosphorylation | IMAC / TiO2 Resins | Physically resolves isobaric positional isomers (e.g., adjacent Serine/Threonine residues) in the gas phase, providing the ultimate physical proof for phosphosite confidence. |
| Ubiquitination | K-ε-GG remnant antibodies | Focuses the concentrated ion beam on specific di-glycine remnants, ensuring that even extremely low-stoichiometry substrates of E3 ligases are captured and quantified accurately. |
| Acetylation | Pan-acetyl antibodies | Effectively mitigates signal loss by pushing low-abundance acetylated peptides above the dense background noise threshold of complex tissue lysates. |
| Methylation | Specific motif antibodies | High-resolution 4D-DIA differentiates subtle mass shifts (such as mono-, di-, and tri-methylation) and positional differences, drastically reducing biological interpretation risk. |
Discovery 4D-PTM vs Targeted PTM Follow-Up: Decision Guide
| Dimension | Standard DDA/DIA PTM | Discovery 4D-PTM | Targeted PTM Follow-Up / 4D-PRM |
|---|---|---|---|
| Isobaric site resolution | Low to Moderate | High (CCS-supported) | Very High (Pre-defined targeting) |
| Enrichment input required | High (mg level) | Low-input compatible (subject to review) | Low to Moderate |
| Typical application | Basic model screening | Deep mechanism & cohort discovery | Clinical biomarker panel validation |
| Verification readiness | Requires heavy filtering | High (Ready for PRM transition) | Absolute (Final clinical proof) |
| Mechanism-readiness | Basic site tables | Network and regulator mapping | Absolute quant of specific pathway nodes |
When broad PTM discovery is the right starting point: If your biological question is entirely open-ended—such as understanding the global cellular regulatory response to a novel therapeutic—a broad Discovery 4D Proteomics workflow is the correct starting point to map the unknown regulatory landscape comprehensively.
When targeted PTM follow-up becomes the better route: Once a comprehensive Discovery project identifies a shortlist of critical regulatory sites, transitioning directly to Targeted Proteomics (such as 4D-PRM) provides the absolute quantification necessary for downstream clinical assay development or functional verification.
Step-by-Step 4D PTM Workflow with Enrichment and QC Checkpoints
Evaluate biological questions and select the optimal modification route.
QC Gate: Pre-enrichment feasibility. Assess available sample volume and matrix type to definitively mitigate workflow risk before lab work begins.
Optimized, inhibitor-protected lysis and modification-specific affinity enrichment.
QC Gate: Post-enrichment specificity. Verify target modification dominance over the peptide pool before proceeding to full acquisition.
TIMS-enabled LC-MS/MS captures vital Collisional Cross Section (CCS) dimension data.
QC Gate: Site localization review. Apply strict thresholds to filter ambiguous assignments and report only highly defensible sites.
Robust data processing pipelines generate rigorous quantitative site matrices.
QC Gate: Batch comparability review. Assess CV distributions of pooled bridge samples to confirm absolute stability across analytical batches.
Sample Requirements for PTM Analysis
| Sample Type | Typical Input | Primary PTM Fit | Main Enrichment Concern |
|---|---|---|---|
| Tissue / fresh frozen | 1-2 mg | Phospho / Ubiquitin / Ac | Endogenous phosphatase/deubiquitinase activity |
| FFPE | 5-15 curls (10μm) | Phospho / Acetylation | Low yield and severe site sparsity due to cross-linking |
| Plasma / serum | 100-300 μL | Glycosylation / Specific Phos | Severe interference from highly abundant background proteins |
| Cells / organoids | 106 - 107 cells | All PTM routes | Biochemical buffer incompatibility (e.g., strong detergents) |
| Low-input clinical | Consult required | Targeted specific PTMs | Near complete sample loss during standard enrichment phases |

Storage and Shipping Considerations: Modifications are highly labile in nature. Immediate flash-freezing and the strict inclusion of PTM-specific inhibitors (e.g., broad-spectrum phosphatase or deubiquitinase inhibitors) during the initial lysis phase are absolutely critical for overall project success and signal preservation.
What You'll Receive: Expected Results and Mechanism-Ready Deliverables
Delivering a basic spreadsheet of modified sites is fundamentally insufficient for modern biology. We explicitly bridge the gap from raw site tables to actionable mechanism hypotheses, structuring our advanced deliverables to support route-specific downstream planning and publication.
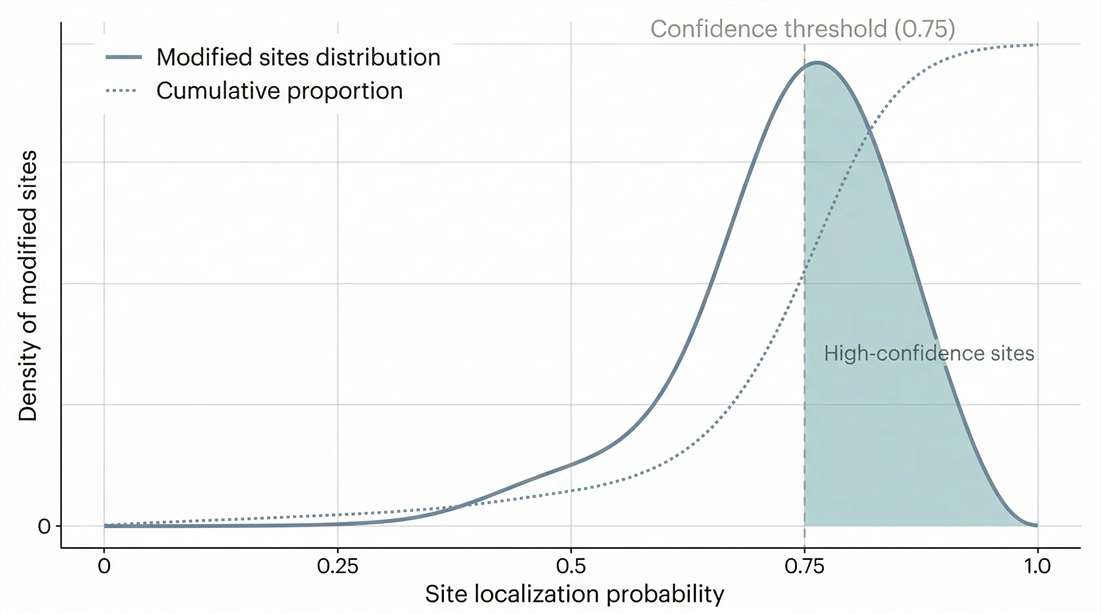
Localization Confidence: Demonstrates the definitive defensibility of the dataset by visualizing the high proportion of sites meeting strict localization probability thresholds.
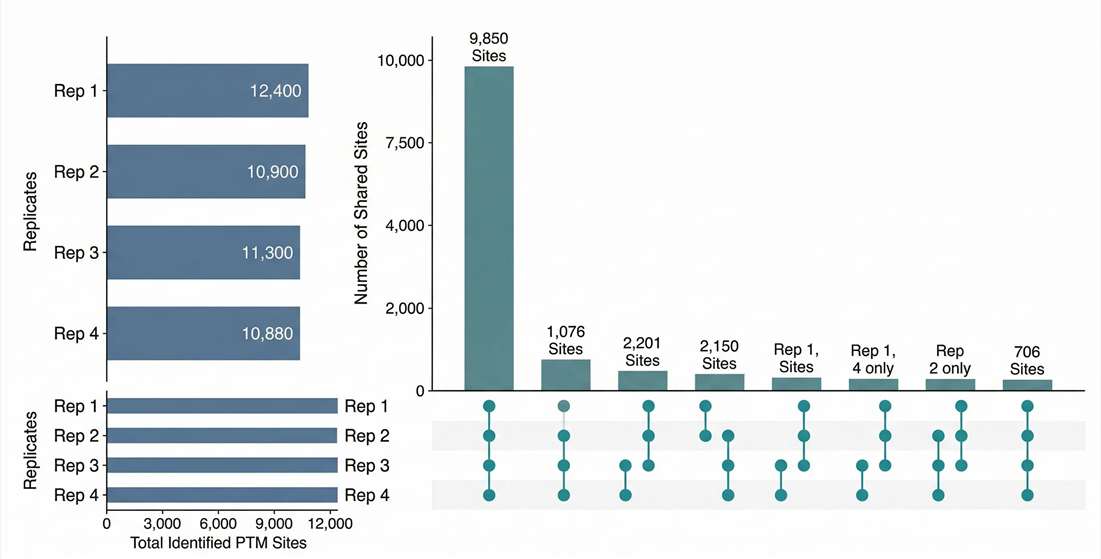
Coverage & Overlap: Visualizes the high specificity of the enrichment process and the quantitative overlap between biological replicates.
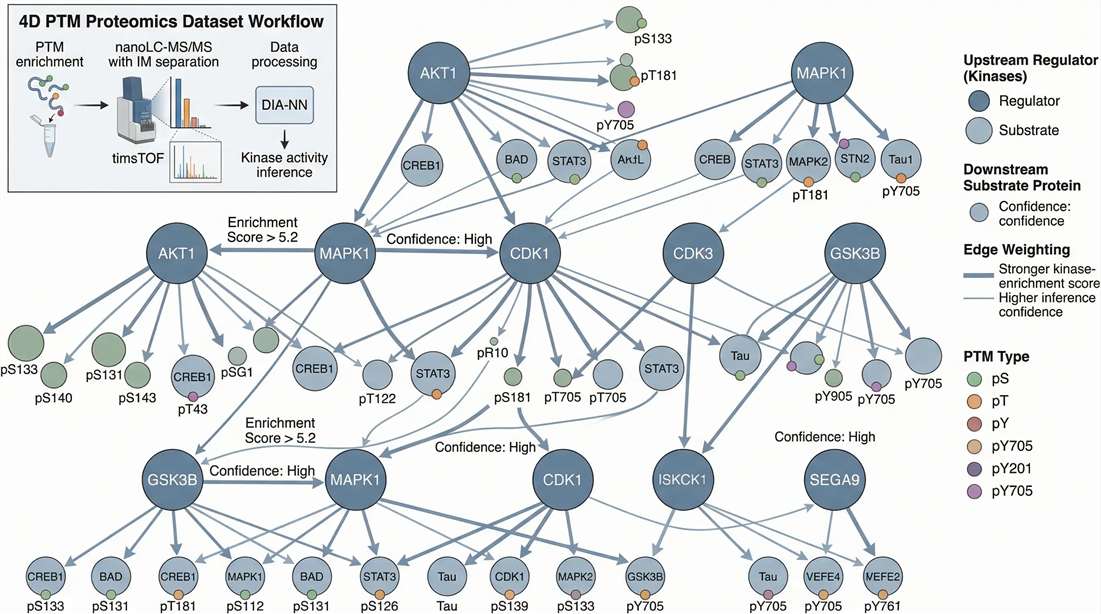
Regulator Network: Shows precisely how your identified modification sites connect to upstream kinases or enzymes (e.g., via KSEA).
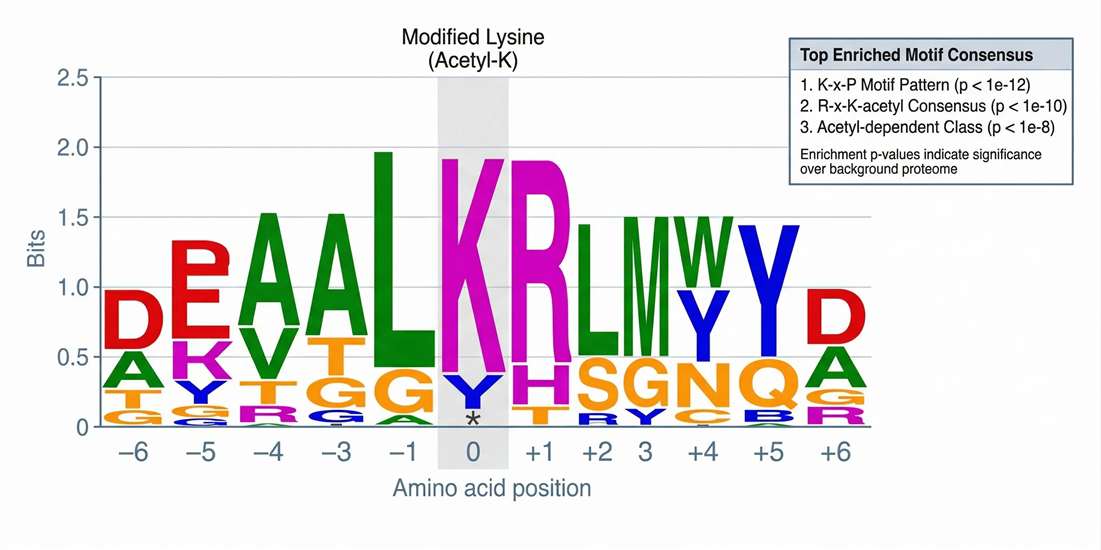
Motif Enrichment: Reveals the highly specific amino acid sequence contexts flanking the modified residues, crucial for upstream enzyme inference.
Through our integrated Proteomics Bioinformatics Analysis Service, we provide a comprehensive, tiered interpretation strategy:
Site Tables with Confidence
- The foundational quantitative data matrix, rigorously filtered for site ambiguity and false discovery control.
Enrichment QC and Completeness Review
- The ultimate quality proof, ensuring that analytical batch effects and background noise are strictly controlled.
Route-Specific Downstream Planning
- Phosphoproteomics: Kinase-substrate enrichment analysis (KSEA) and highly precise upstream kinase inference.
- Ubiquitination Proteomics: Degradation logic mapping and targeted E3 ligase substrate inference.
- Acetylation Proteomics: Chromatin state modeling and deep transcription factor activity mapping.
- Methylation Proteomics: Regulator-state interpretation and complex epigenetic pathway modeling.
Next-Step Validation Support
- Our reports highlight the highest-confidence regulatory nodes for targeted 4D-PRM follow-up.
Representative Evidence for 4D-PTM Proteomics
Hybrid DDA/DIA-PASEF Enables Deep Proteotyping of Triple-Negative Breast Cancer
Journal: Scientific Data · Published: 2024
Study Scope
Researchers developed a hybrid DDA/DIA-PASEF assay library to enable deep proteotyping of triple-negative breast cancer (TNBC), a biologically aggressive and heterogeneous tumor subtype that requires broad and reproducible proteome coverage for molecular characterization.
- Proteins were extracted from 105 TNBC tissues and used to generate a TNBC-specific assay library.
- Pooled samples were fractionated and analyzed in DDA-PASEF mode on a timsTOF Pro LC-MS system.
- 16 individual TNBC lysates were analyzed in DIA-PASEF mode to demonstrate real-sample applicability.
- The study compared library-based and library-free DIA data extraction strategies in Spectronaut and DIA-NN.
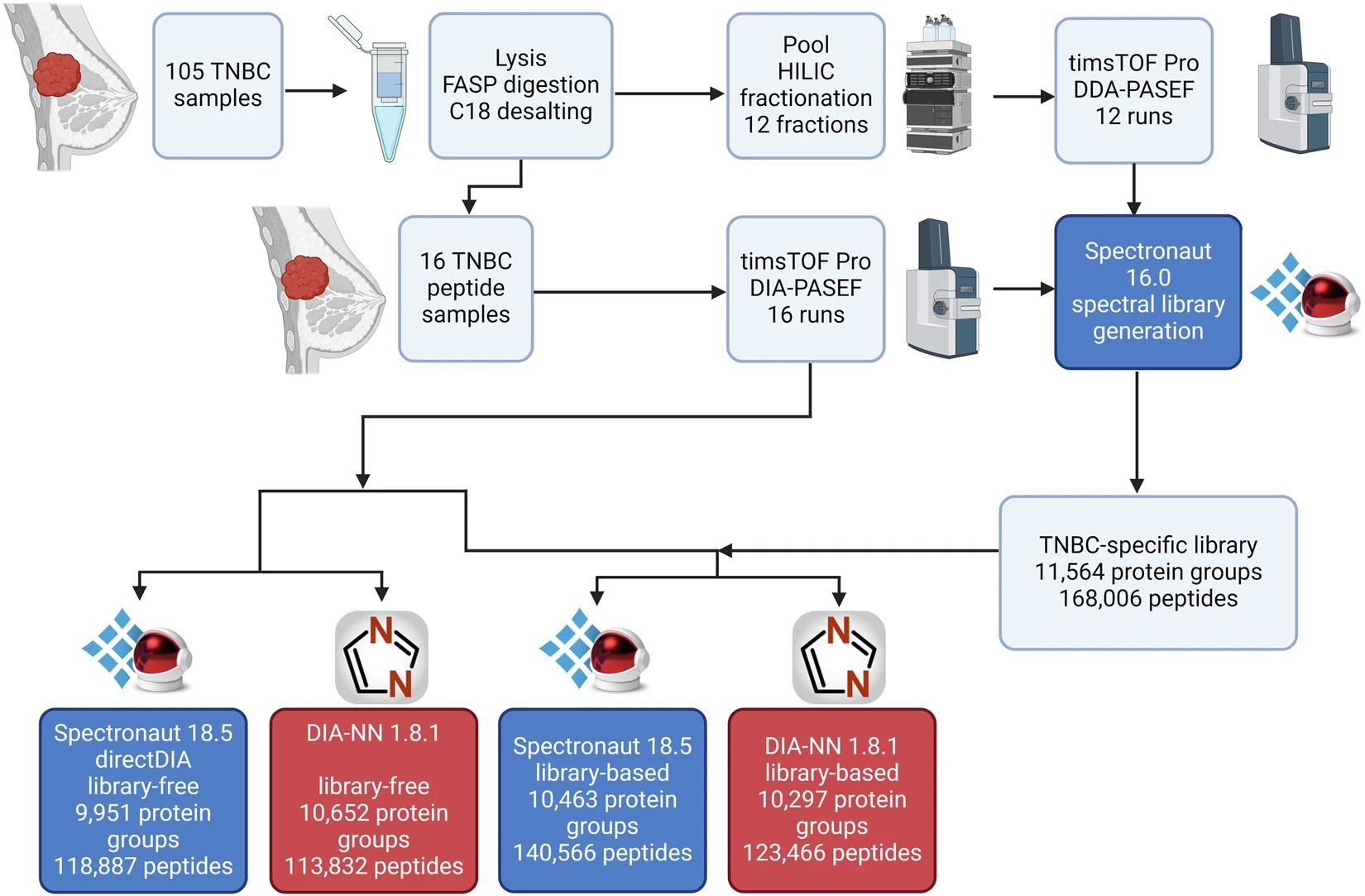 Workflow of the study.
Workflow of the study.
Proteomic Coverage and Data Quality
- The hybrid TNBC assay library covered 244,464 precursors, 168,006 peptides, and 11,564 protein groups at 1% FDR.
- When applied to 16 individual TNBC samples, the library-based workflow increased identifications compared with library-free processing.
- In Spectronaut 18.5, the best-performing setup reported 190,310 precursors, 140,566 peptides, and 10,463 protein groups.
- The application results also showed sample clustering, intensity behavior, missing values, completeness, and coefficient-of-variation distributions suitable for assessing quantitative performance.
These results demonstrate how DDA/DIA-PASEF can improve proteome depth while preserving the structured quantitative outputs needed for biological interpretation and downstream decision-making in complex tissue studies.
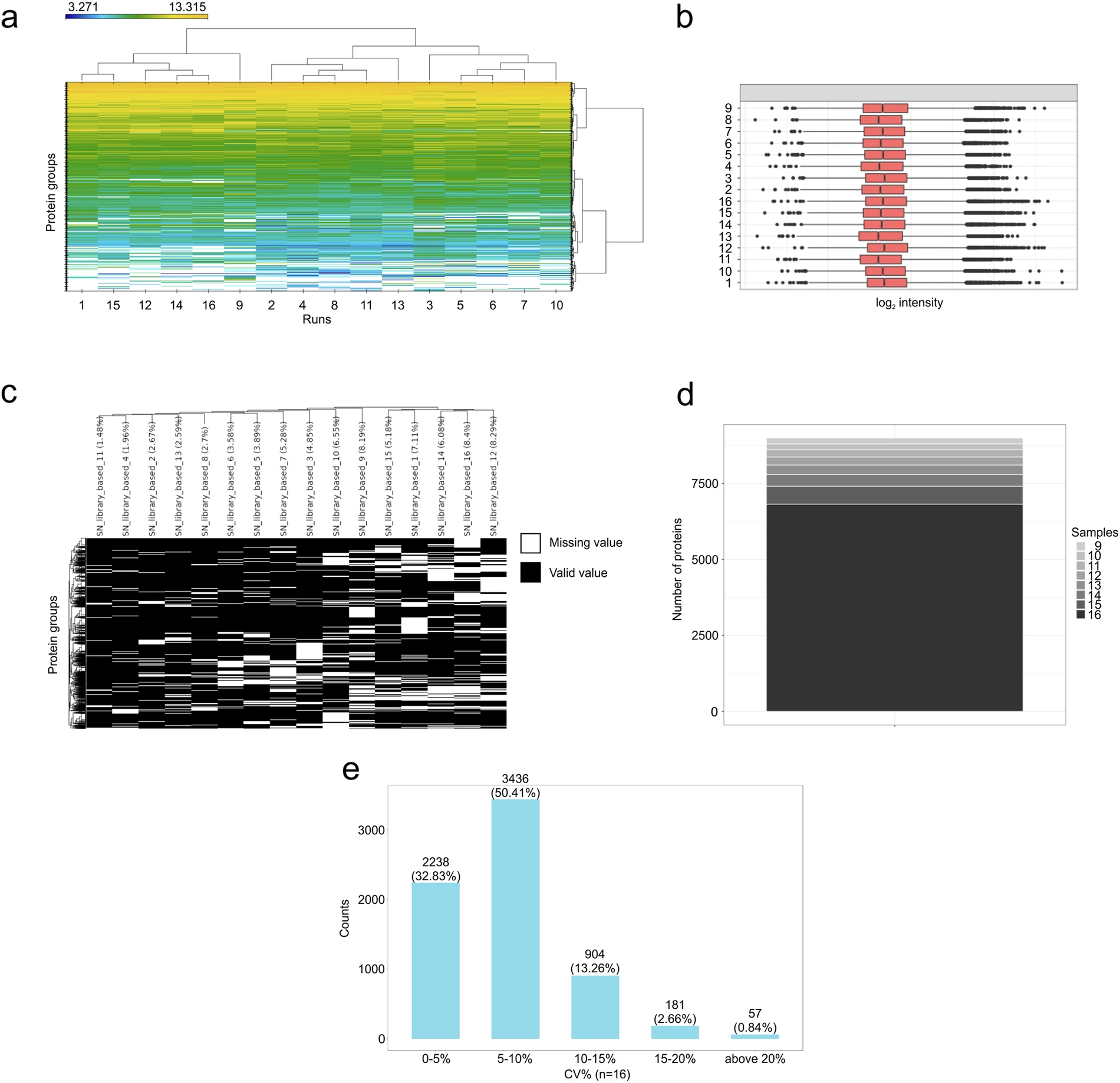 DIA-PASEF application to TNBC samples demonstrates quantitative coverage and data-quality characteristics.
DIA-PASEF application to TNBC samples demonstrates quantitative coverage and data-quality characteristics.
Biological Insights
This study supports several practical conclusions for deep modification-aware proteomics:
- A TNBC-specific hybrid assay library can substantially improve proteome depth in a difficult disease matrix.
- DIA-PASEF can be applied to individual TNBC tissue samples with strong quantitative completeness and consistency.
- The resulting dataset structure supports subgroup comparison, candidate prioritization, and downstream biological interpretation in cancer-focused proteomics studies.
Technical Highlights
- Large-scale assay-library generation from pooled TNBC tissue material.
- DDA-PASEF for library expansion combined with DIA-PASEF for individual-sample quantification.
- Direct comparison between library-based and library-free extraction strategies.
- Quantitative outputs include clustering, missing values, completeness, and CV-based precision review.
Why It Matters
This study is a strong fit for a 4D-PTM parent page because it connects deep assay-library construction with real tissue application. It shows how PASEF-based workflows can support deeper coverage and robust quantitative interpretation in a clinically relevant, high-complexity matrix while preserving the result types decision-makers care about most: completeness, reproducibility, and interpretability.
Reference
Lapcik, Petr, et al. "A hybrid DDA/DIA-PASEF based assay library for a deep proteotyping of triple-negative breast cancer." Scientific Data 11, 794 (2024).