Large-scale DIA Phosphoproteomics Quantification In Large Samples
Difficulties In Reproducibly Analyzing Phosphorylation Sites In Large Samples
Site-specific protein phosphorylation is one of the most important post-translational modifications, and dysregulated phosphorylation signaling is a hallmark of cancer and many other diseases. However, most phosphoproteomic studies require measurement by LC-MS/MS for days or even weeks, analyzing a small subset of cells under specific conditions to identify functionally relevant phosphorylation sites. Furthermore, current systems for reproducibly analyzing phosphorylation sites in large numbers of samples remain challenging, which limits high-throughput applications of phosphoproteomics, such as drug screening.
How To Solve The Problem Of Reproducible Analysis Of Phosphorylation Sites In Large Samples
To address these issues, Jesper V. Olsen's team published a research paper titled "Rapid and site-specific deep phosphoproteome profiling by data-independent acquisition without the need for spectral libraries" in Nature Communications. This study optimized the data analysis workflow for DIA phosphoproteomics and systematically analyzed the effects of kinase inhibitors on the phosphoproteome. The authors developed an optimized untargeted phosphoproteomics approach that combines LC-MS/MS with DIA. This approach enables systematic and reproducible analysis of over 10,000 phosphorylation sites in hundreds of samples. In addition, the authors developed and used algorithms to accurately locate phosphorylation sites in the DIA dataset and determine their stoichiometry on a system-wide scale. This technique was applied to identify phosphorylation site targets for ten major protein kinases in the epidermal growth factor signaling pathway.
Comparing DDA and DIA quantification For Phosphoproteomics
In order to perform high-throughput phosphoproteomics research, it is necessary to reduce the amount of input protein, improve the reproducibility of the workflow, and minimize the usage time of mass spectrometers. Therefore, phosphopeptides enriched from 200μg starting trypsin-digested material by high-throughput magnetic Ti-IMAC beads were optimized for a scalable single analysis workflow. A fast 28 Hz high-energy collision dissociation (HCD) scan method on a Q Exactive HF-X mass spectrometer was used to routinely quantify ~7000 phosphopeptides within 15 minutes of LC-MS/MS analysis time, with an overall identification rate of over 50%.
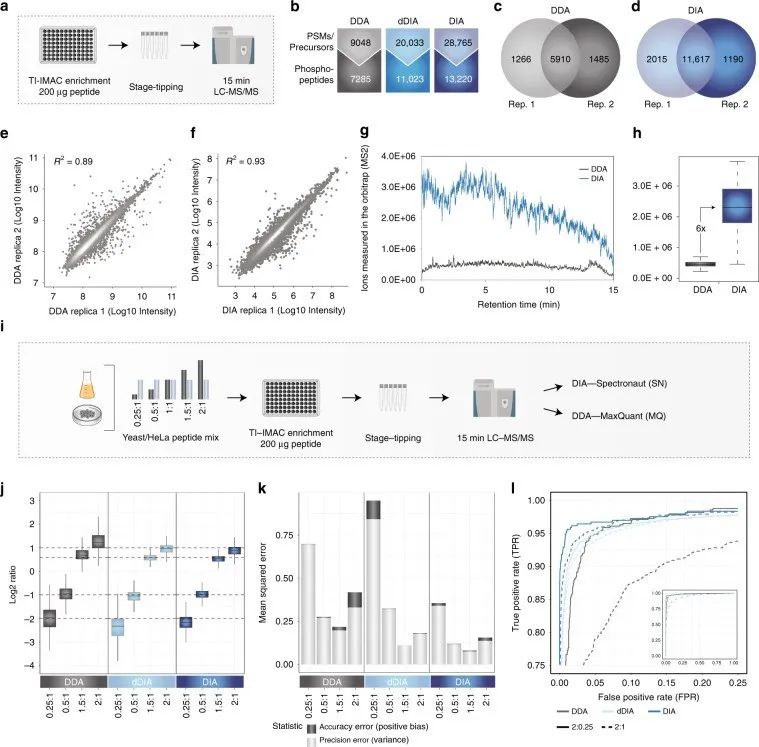 Figure 1 | Identification and quantification of phosphorylated proteins using DDA and DIA phosphoproteomics
Figure 1 | Identification and quantification of phosphorylated proteins using DDA and DIA phosphoproteomics
a. Workflow of phosphoproteomics experiment;
b. Comparison of DDA and DIA quantification of phosphopeptides;
c. d. Overlap of phosphopeptides between two DDA/DIA replicates;
d. f. Correlation between DDA/DIA replicates;
g. Measurement of ions in the ion trap by DDA and DIA in MS2 scan;
h. Quantitative differences of DDA and DIA ions measured in the ion trap;
i. Workflow of experiment.
Additionally, the authors optimized instrument settings by adjusting parameter settings to achieve optimal DIA performance. They calculated mean squared error (MSE) as the sum of bias and variance for each method, representing the quantitative error in accuracy and precision, respectively, to better evaluate quantitative accuracy. They used significance d-scores from SAM tests to calculate true positive rate (TPR) and false positive rate (FPR) for DIA, in order to test whether accurate and precise quantification with DIA can better identify targets.
1. DIA-specific Phosphorylation Site Localization Algorithm
The authors developed a PTM localization algorithm for peptide analysis using information not available in standard DDA data. This includes complete isotope pattern of fragment ions and the possibility of generating short chromatograms correlated with target precursor peaks, which systematically removes any interfering fragment ions that cannot be resolved in DDA. Combining these two aspects with other scores based on fragment ion intensity and mass accuracy, specific weighted scores for each fragment are calculated and used to compute specific site localization scores.
2. Comparison of DDA and DIA In Biological Environment
Next, the authors evaluated whether the increased coverage of local phosphorylated peptides in DIA compared to DDA translates into an advantage in cellular signaling studies. They used retinal pigment epithelial cells (RPE1) stimulated with EGF and treated with different MEK kinase inhibitors as a model system and analyzed the samples using 15-minute gradients with both DDA and DIA methods.
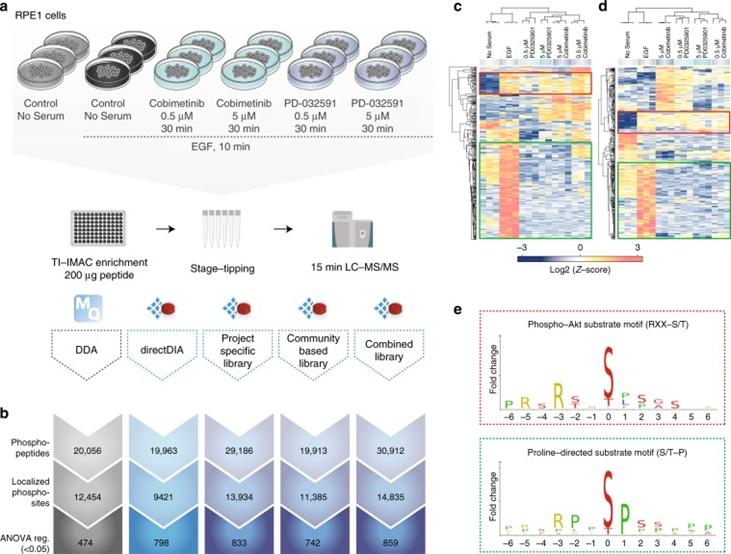 Figure 2 | A comparison of DDA and different types of DIA in biological settings.
Figure 2 | A comparison of DDA and different types of DIA in biological settings.
a. Experimental workflow; b. Overview of different methods for identifying phosphopeptides, localized phosphosites, and ANOVA-regulated phosphosites; c. DDA; d. Heatmap of clustering analysis of DIA phosphorylation sites; e. Motif analysis.
3. Analysis Of Phosphorylation Site Stoichiometry
In addition to the relative quantification of phosphorylation sites, determining their occupancy or absolute stoichiometry is also valuable. The combination of high-scoring chemometrics and dynamic regulation is strong evidence for the site's function in the cellular environment under investigation. It is possible to determine the stoichiometry of large-scale phosphorylation sites by using the proportion observed between the two phosphopeptides, their corresponding unphosphorylated peptides, and the corresponding protein from SILAC data processing conditions, as well as TMT-reuse data.
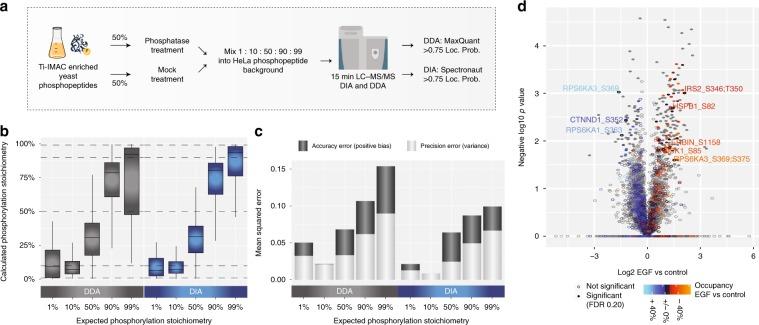 Figure 3 | Workflow for the control ratio experiment.
Figure 3 | Workflow for the control ratio experiment.
4. Large-scale Phosphoproteomics Using Kinase Inhibitors
To demonstrate the power and scalability of the developed DIA-based fast site-specific phosphoproteomics workflow, the authors applied it to the phosphorylation site targets of ten major protein kinases in the epidermal growth factor signaling pathway using 30 kinase inhibitors.
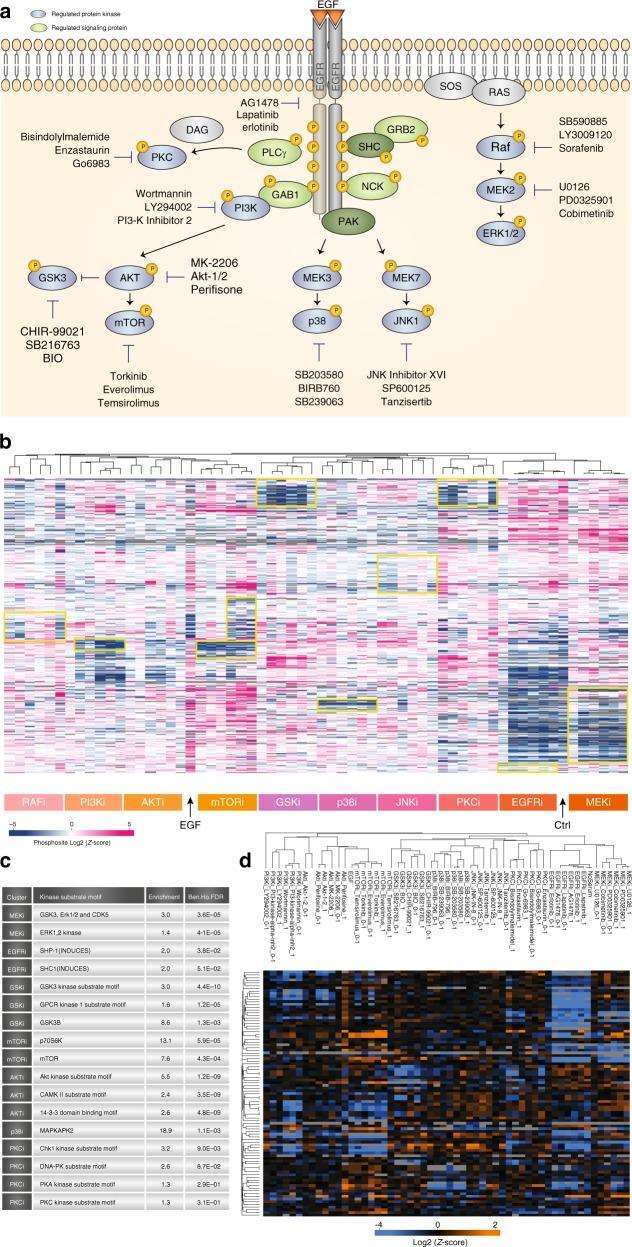 Figure 4 | Kinase Inhibitor Screening
Figure 4 | Kinase Inhibitor Screening
a. Overview of Kinase Inhibitor Experiment; b. Hierarchical clustering of mean site intensity measurements; c. Fisher's exact test for over-represented kinase motifs; d. Clustering analysis of known substrates and individual kinases.
Conclution
The authors optimized the workflow of phosphoproteomics based on DIA, and developed a fast and reproducible method to independently analyze hundreds of phosphorylated proteins using DIA analysis. Compared to the currently advanced data-dependent acquisition (DDA) based phosphoproteomics, DIA-based phosphoproteomics has a wider dynamic range, higher identification reproducibility, and higher quantitative sensitivity and accuracy. They also developed a PTM localization algorithm as part of the DIA calculation, which served as a benchmark for phosphoproteomics. The specificity and high quality of DIA-based phosphoproteomics were demonstrated through the analysis of synthetic phosphopeptides. Additionally, kinase inhibitors' effects on phosphoproteomics were analyzed using a DIA-based label-free quantification technique.
Reference:
- Jesper V. Olsen et al., Rapid and site-specific deep phosphoproteome profiling by data-independent acquisition without the need for spectral libraries Nature communications(2019)

 4D Proteomics with Data-Independent Acquisition (DIA)
4D Proteomics with Data-Independent Acquisition (DIA)