- Services
- FAQ
- Demo
- Case Study
- Related Services
- Support Documents
- Inquiry
What is Circular Dichroism (CD) Spectroscopy
Since the pioneering work of Kendrew, who utilized X-ray technology to unveil the folded structure of myoglobin, protein research has evolved from the determination of amino acid sequences to the in-depth exploration of spatial structures and conformations. It has become increasingly apparent that understanding the conformation of proteins and their related variations is of paramount significance for comprehending their physiological functions.
Circular dichroism (CD) spectroscopy has emerged as a rapid, straightforward, and reasonably accurate method for the investigation of protein conformations. In 1969, Greenfield was among the pioneers in utilizing CD spectroscopic data for estimating protein conformations, and subsequent research methodologies have been consistently documented. Significantly, over the last two decades, the analysis of protein secondary structures using far-ultraviolet CD (Far-UV CD) data has experienced notable advancements. These advancements encompass not only computational methods and fitting procedures but also coincide with the progress in X-ray crystallography and nuclear magnetic resonance techniques. Consequently, this progress has facilitated the precise determination of protein conformations, enhancing the accuracy of CD data fitting through more comprehensive databases.
Furthermore, researchers have unveiled distinctive advantages in the exploration of protein tertiary structures through CD spectroscopy. Specialized methods and accompanying software have been devised for the identification of protein tertiary structures employing far-ultraviolet CD spectroscopy. Additionally, near-ultraviolet circular dichroism (Near-UV CD), renowned for its sensitivity as a spectral probe, has the capacity to discern alterations in the microenvironment surrounding aromatic amino acid residues and disulfide bonds within proteins. Consequently, CD spectroscopy, serving as an indispensable instrument for the examination of protein or peptide conformation in solution, has engendered considerable interest among the scientific community.
At Creative Proteomics, we specialize in delivering advanced CD Spectroscopy services aimed at comprehensive protein structure elucidation. With decades of experience in the biotechnology industry, our team of experts utilizes state-of-the-art technology to provide precise, reliable, and insightful analyses of protein structures.
The Principles of CD Spectroscopy
CD spectroscopy is based on the differential absorption of left- and right-circularly polarized light by chiral molecules. When a protein sample is exposed to plane-polarized light, its optically active components—such as peptide bonds, aromatic amino acids, and disulfide bridges—absorb the polarized light differently. This differential absorption generates a characteristic CD spectrum, which provides direct information about the molecule's chiral properties.
CD spectroscopy is typically divided into two key spectral regions, each offering distinct insights into different aspects of protein structure:
Far-UV CD Spectroscopy (190-250 nm): This region primarily probes the secondary structure of proteins, encompassing the repetitive patterns of folding such as α-helices, β-sheets, and random coils. Each of these structural motifs exhibits a characteristic CD signature. For instance, the α-helix generates pronounced negative bands near 222 nm and 208 nm, with a positive band around 190 nm. Analysis of these signals enables to quantify the relative proportions of various secondary structure elements, offering critical insights into protein folding and stability.
Near-UV CD Spectroscopy (250-350 nm): In the near-UV region, CD spectroscopy targets the tertiary structure of proteins, particularly focusing on the microenvironment surrounding aromatic side chains (e.g., phenylalanine, tyrosine, and tryptophan) and disulfide bonds. Changes in the near-UV CD spectrum often reflect perturbations in the tertiary structure, enabling the study of protein folding, conformational transitions, and protein-ligand interactions.
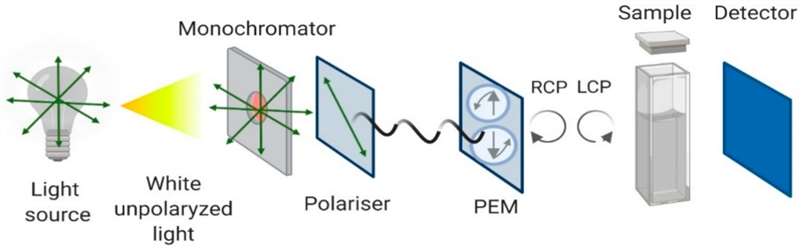 Figure 1. Schematic Representation of CD Principles
Figure 1. Schematic Representation of CD PrinciplesFor more information see our detailed knowledge on circular dichroism spectroscopy.
We Can Provide but Not Limited to
- Protein Secondary Structure Determination: CD spectra in the far ultraviolet region provide information about the arrangement of peptide bonds within proteins, allowing the calculation of the proportions of protein secondary structures such as α-helices, β-sheets, turns, and irregular coils.
- Protein Tertiary Structure Characterization: CD spectra in the near-ultraviolet region reveal the distribution of amino acid residues with chromophoric side chains, including tryptophan, phenylalanine, tyrosine, and variations in the disulfide bond environment.
- Investigation of Thermal Denaturation and Thermodynamic Parameters: Thorough studies of thermal denaturation processes using temperature-controlled CD spectrophotometry to determine Tm values, crucial for evaluating protein stability.
- Protein Stability Studies: Monitoring of protein folding, unfolding, and denaturation processes under various environmental conditions.
- Protein Interaction Analysis: Examination of protein-protein and protein-nucleic acid interactions through conformational changes.
CD Spectroscopy Analysis Workflow
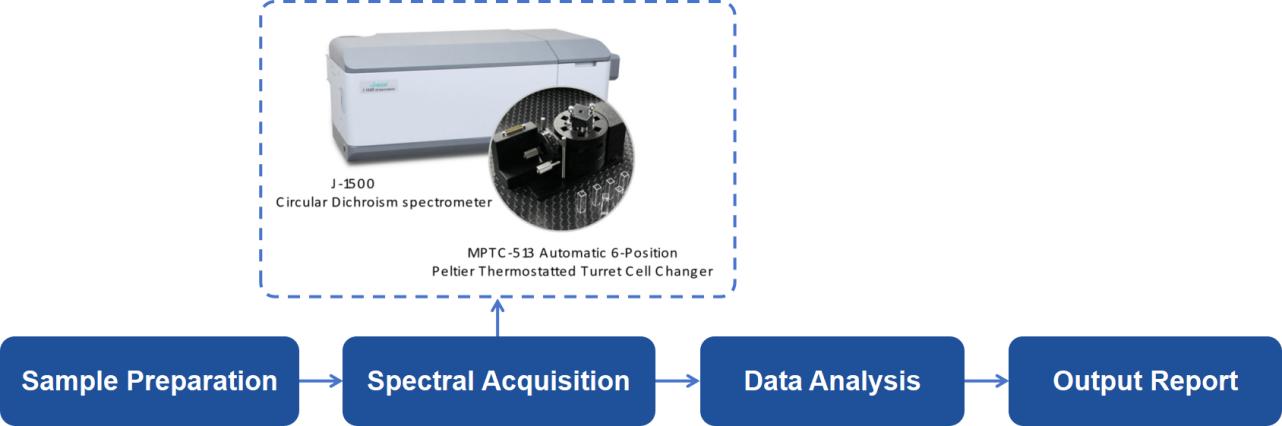
At Creative Proteomics, our CD spectroscopy analysis platform is equipped with cutting-edge technology to ensure precise and comprehensive results. Our workflow includes the following steps:
Sample Preparation: We accept protein and peptide samples in various forms, including liquid solutions and dry powders, ensuring flexibility in research needs.
Spectral Acquisition: Using Jasco J-1500 spectropolarimeter equipped with Peltier temperature controller to capture both far-UV and near-UV spectra to analyze secondary and tertiary structures, respectively.
Data Analysis: We apply sophisticated spectral deconvolution methods to extract quantitative data on secondary and tertiary structure composition.
Comprehensive Reporting: Our expert team provides detailed reports, including visual spectra, quantitative data on protein conformations, and interpretations relevant to your research.
Sample Requirements
- Sample Types: Protein, nucleic acid samples
- Sample Forms: Liquid, dry powder
- Sample Volume: Minimum sample volume required is generally 200 µL for liquid samples.
- Purity: Samples should be as pure as possible to avoid interference in spectral analysis.
- Buffer Compatibility: Use buffers compatible with CD spectroscopy (e.g., phosphate, acetate). Avoid strong absorbents like Tris or high concentrations of salt.
- Temperature Stability: Samples should be stored at low temperatures (dry ice) until analysis to maintain stability and prevent degradation.
Please ensure that all samples are labeled clearly and submitted with a completed submission form detailing any specific requirements or conditions for analysis.
FAQ
Q: What information do circular dichroism spectra obtained in the near-ultraviolet and far-ultraviolet regions convey?
A: Circular dichroism spectra in the far-ultraviolet region offer insights into the secondary structure of the protein. These spectra illuminate the spatial orientation of peptide bonds within the protein, facilitating the calculation of the proportions of distinct secondary structural elements, such as α-helices, β-sheets, turns, and irregular coils. This data is pivotal for a comprehensive understanding of the protein's structural attributes and stability.
Conversely, circular dichroism spectra in the near-ultraviolet region provide details regarding the protein's side chains. They depict the spatial arrangement of amino acid residues containing chromophoric side groups, such as tryptophan, phenylalanine, and tyrosine, within the protein's structure. Furthermore, near-ultraviolet scans can unveil alterations in the microenvironment of disulfide bonds within the protein, a valuable resource for the investigation of the protein's functional attributes and interactions.
Q: What software is employed for the analysis of protein secondary structure, and what are its merits?
A: Currently, there exist multiple software alternatives for the analysis and prediction of protein secondary structure. Among these, CDNN stands out as a secondary structure prediction software rooted in deep neural network technology. Its capabilities encompass the prediction of a protein's secondary structure composition through the analysis of its amino acid sequence.
CDNN presents several advantages when compared to other software solutions:
High Precision: CDNN yields remarkably precise calculations, facilitating accurate predictions of a protein's secondary structure.
Expediency: CDNN boasts swift computation capabilities, enabling the expeditious processing of extensive protein sequence datasets.
Cost Efficiency: CDNN offers a cost-effective solution, resulting in savings in both analysis costs and time.
Q: Dose the types of buffers affect CD spectroscopy?
A: Yes, it's essential to use buffers that do not absorb significantly in the UV range, typically below 220 nm. Commonly used buffers include phosphate-buffered saline (PBS) and Tris buffer, but it's best to verify the compatibility of your specific buffer with the CD analysis.
Demo
Demo 1: Characterization of Protein Higher-Order Structures The secondary structure of protein samples
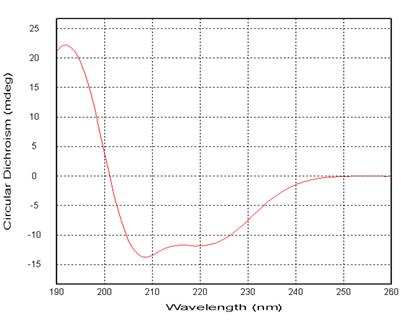 Figure 2: Far Ultraviolet Circular Dichroism Spectrum of Protein
Figure 2: Far Ultraviolet Circular Dichroism Spectrum of Protein| Structure | Proportions |
|---|---|
| Helix | xx.9% |
| Antiparallel | x.0% |
| Parallel | x.5% |
| Beta-turn | xx.2% |
| Random Coil | xx.4% |
| Total Sum | 100% |
Table 1: CDNN Software for Protein Secondary Structure Prediction (190nm-260nm) Protein Sample Tertiary Structure
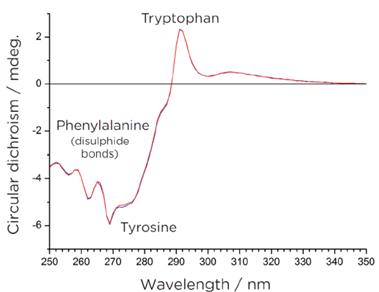 Figure 3: Near Ultraviolet Circular Dichroism Spectrum of Proteins
Figure 3: Near Ultraviolet Circular Dichroism Spectrum of ProteinsThis spectral profile captures the absorption characteristics of aromatic amino acid residues, including tryptophan, phenylalanine, and tyrosine, and also reveals changes in the microenvironment of disulfide bonds. The influence of the surrounding environment imparts nuanced circular dichroism signals within this specific wavelength range. Monitoring alterations in these signals offers valuable insights into modifications in the protein's tertiary structure.
Demo 2: Characterization of Nucleic Acid Higher-Order Structures
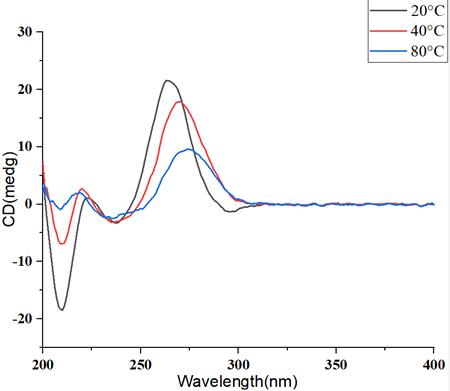 Figure 4: Circular Dichroism Spectra of mRNA at Different Temperature Gradients
Figure 4: Circular Dichroism Spectra of mRNA at Different Temperature GradientsWith the elevation of temperature, the higher-order structure of mRNA undergoes disruption, resulting in a pronounced decline in the circular dichroism signal, accompanied by a concurrent shift in the peak position.
Case Study
Case: In Vitro Functional Quality Characterization of NOTA-Modified Somatropins
Background
The regulatory landscape for biopharmaceuticals is shifting toward more comprehensive functional characterization, beyond traditional physicochemical evaluation. Techniques like SEC and MS, typically used to analyze molecular mass and modifications, are increasingly utilized to assess the biological functionality of proteins, including their binding affinity to receptors. Somatropin, a human growth hormone analog, was modified with NOTA, and the study aimed to assess how these modifications affected its functional binding and biological activity.
Methods
- Size Exclusion Chromatography (SEC)/High-Performance Receptor-Binding Chromatography (HPRBC): These techniques were used to analyze the formation of somatropin–hGHBp complexes, identifying the ratios of receptor binding (2:1 or 1:1) based on molar excess conditions.
- Native Mass Spectrometry (MS): Native MS was employed to analyze the binding of NOTA-modified somatropin to hGHBp, providing insights into the substitution degree and structural changes in the protein.
- Circular Dichroism (CD): CD spectroscopy was used to examine conformational changes in somatropin, especially in its native-like conditions.
- Biosensor Assays (Surface Acoustic Wave (SAW) technology): SAW biosensors evaluated the binding kinetics of somatropin and NOTA-modified variants, confirming that the binding affinity to antibodies remained unchanged.
- Cell Assays: A classical bioassay involving the growth hormone receptor (GHR) and the JAK2 signaling pathway was conducted to measure the biological efficacy and potency of the modified somatropin.
Results
- Binding Affinity: CucE exhibited a higher binding affinity for HSA compared to CucI, with an average dissociation constant (Kd) of 5.0 × 10⁻⁷ M for CucE and 1.5 × 10⁻⁶ M for CucI.
- Kinetic Analysis: The association rates (kon) were found to be significantly faster for CucE than CucI, indicating that CucE's higher affinity is partly due to its quicker binding kinetics.
- CD Analysis: Both cucurbitacins displayed similar CD signals, indicating non-stereospecific binding to serum albumins. Upon interaction with HSA, the intensity of the positive CD band of biliverdin (an indicator of binding) increased, suggesting allosteric modulation. In contrast, when tested with RSA, the same band's intensity decreased, implying potential negative allosteric modulation.
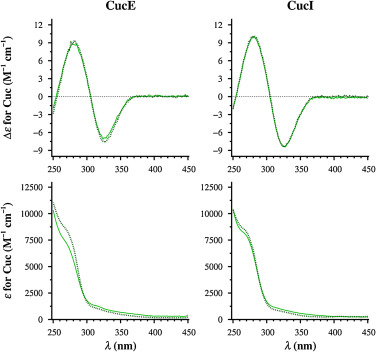 Figure 5. CD and UV spectra of CucE and CucI in solution (solid lines) and protein-corrected spectra of their 2:1 complexes with HSA (dotted lines).
Figure 5. CD and UV spectra of CucE and CucI in solution (solid lines) and protein-corrected spectra of their 2:1 complexes with HSA (dotted lines).References
- Yao H, et al. Circular dichroism in functional quality evaluation of medicines. Journal of Pharmaceutical and Biomedical Analysis, 2018, 147: 50-64.
- Pignataro M F, et al. Evaluation of peptide/protein self-assembly and aggregation by spectroscopic methods. Molecules, 2020, 25(20): 4854.
- Li Z, Hirst J D. Quantitative first principles calculations of protein circular dichroism in the near-ultraviolet. Chemical science, 2017, 8(6): 4318-4333.
- Greenfield N J. Circular dichroism (CD) analyses of protein-protein interactions//Protein-Protein Interactions. Humana Press, New York, NY, 2015: 239-265.
- Edoardo Fabini, et al. Surface plasmon resonance and circular dichroism characterization of cucurbitacins binding to serum albumins for early pharmacokinetic profiling. Journal of Pharmaceutical and Biomedical Analysis, 2016, 122: 166-172,
Related Services

Near UV Circular Dichroism Spectroscopy Analysis Service
Accessing near UV circular dichroism spectroscopy analysis services from Creative Proteomics, all delivered in accordance with GLP/GMP standards.

Far UV Circular Dichroism Spectroscopy Analysis Service
Creative Proteomics offers customers far UV circular dichroism spectroscopy analysis services in compliance with GLP/GMP standards.

Advanced Structure Analysis Service
Creative Proteomics employs advanced techniques such as spectroscopy, mass spectrometry, GC/MS, LC/MS, and CE/MS to analyze the structure and stability of your biopharmaceutical formulation.
Support Documents



KNOWLEDGE CENTER

KNOWLEDGE CENTER





