- Services
- FAQ
- Case Study
- Related Services
- Support Documents
- Inquiry
What is HDX-MS Epitope Mapping
Epitopes are the basis of protein antigenicity, and the study of protein antigenic epitopes is important for the design of novel vaccine molecules, novel diagnostic reagents, peptides, therapeutic antibodies, etc. with immunogenic and neutralizing activity. Creative Proteomics offers a comprehensive epitope mapping service, employing advanced techniques like hydrogen-deuterium exchange mass spectrometry (HDX-MS) and cross-linking mass spectrometry (XL-MS), peptide scanning and X-ray co-crystallization to address various epitope types, ensuring accurate and actionable results for vaccine and drug development.
HDX-MS, which has emerged at the forefront of protein research, can provide direct information on which amino acid sequences are located at the surface positions of protein spatial structures (including those under dynamic change), possible active sites and protein-protein interaction sites.
More comprehensive epitope structure information for antigen-antibody interaction studies epitope recognition with functional mAbs is critical for understanding the nature of the immune response and improving the design of vaccines, therapeutics, and diagnostics. In recent years, the identification of B cell epitopes targeting neutralizing antibodies has facilitated the design of peptide-based vaccines against highly mutated pathogens such as HIV, respiratory syncytial virus, and Helicobacter pylori. But these products have not yet entered the clinical stage. Conventional linear epitope recognition can only partially reflect the immunogenicity of the epitope in its native conformation. HDX-MS can quickly provide comprehensive information on epitope structure.
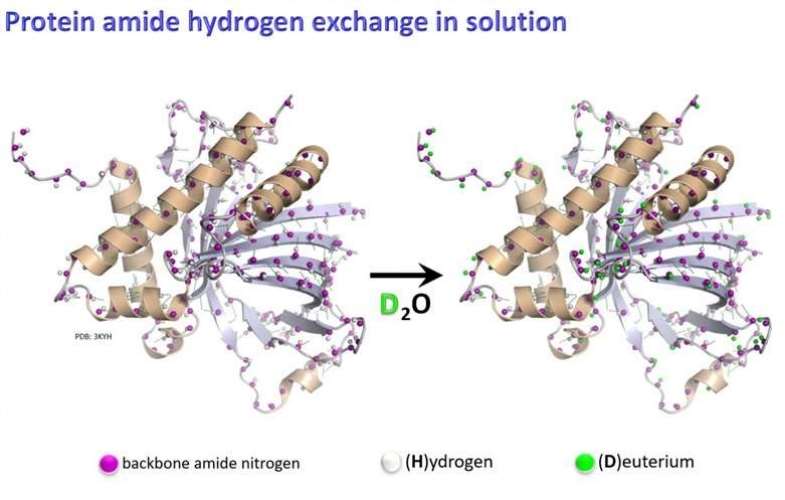 Figure 1. HDX-MS for protein dynamic structure research (John Engen, et al., 2012)
Figure 1. HDX-MS for protein dynamic structure research (John Engen, et al., 2012)Applications of HDX-MS Epitope Mapping
More rapid identification of antigenic sites for drug screening of influenza virus hemagglutinin antigenic epitopes
Epitope characterization is critical for resolving the mechanism of action of drug candidates. Traditional X-ray crystal diffraction methods are often time consuming to collect data from protein crystals. Due to the intrinsic complexity of proteins and the difficulty in obtaining ideal single crystals for specific manipulations, this approach is not easily successful and is not realistic when large numbers of drug candidates need to be screened to identify similar targets. Nuclear magnetic resonance (NMR) techniques are often limited by the molecular weight of the protein.
The Janssen Vaccine and Prevention Research Center used HDX-MS to develop the conformational effects of different drug molecules combined with hemagglutinin HA. Based on the HDX-MS platform, it systematically explored the combination of HA subtypes with 4 different drugs (including monoclonal antibody and small molecule synthetic peptide) candidates. This rapid, low-cost HDX-MS epitope method accurately identified all major antigenic sites.
This screening strategy offers the following advantages in early drug discovery:
- Low sample volume requirements
- Short collection and analysis time
- No molecular weight restrictions
- Protein dynamics data in the natural environment
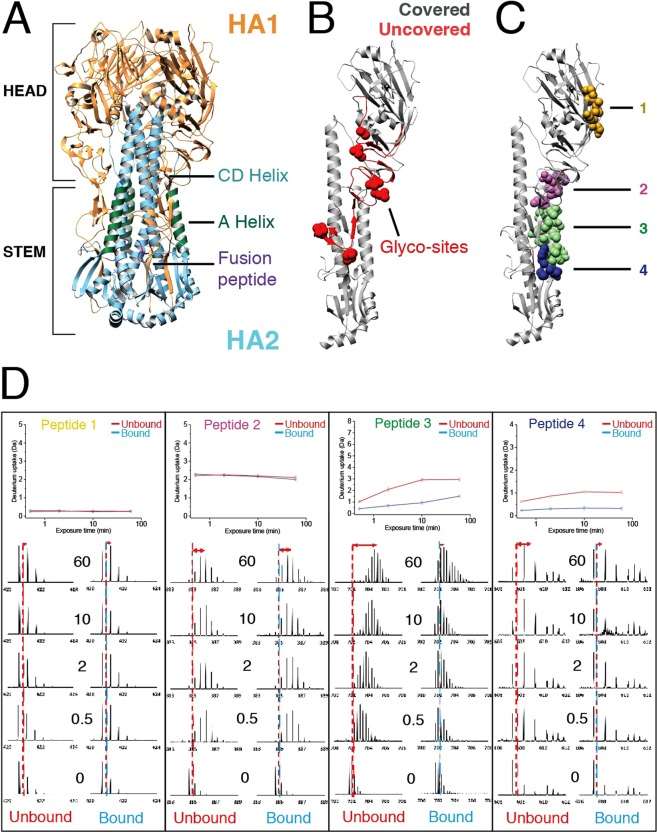 Figure 2. HDX-MS reveals the conformational dynamics of free- and Fab-bound HA trimers in solution (Cristina Puchades, et al., 2019)
Figure 2. HDX-MS reveals the conformational dynamics of free- and Fab-bound HA trimers in solution (Cristina Puchades, et al., 2019)More accurate target identification for antibody-virus particle complex epitope studies
An in-depth understanding of virus structure and surface antigens can reveal the molecular mechanism of virus infection of cells and drug action, which is the basis for research on virus prevention and drug development. HDX-MS is also widely used in virus structure research, including virus epitope identification, virus capsid folding, aggregation, assembly and maturation, virus structure changes and dynamics, virus-cell membrane fusion, etc.
Emerging tools for protein structure analysis
As a method for analyzing protein structures and their molecular dynamics, HDX-MS is mainly used to help scientists gain insight into structural changes in proteins. This technique has a wide range of applications in the study of antigen-antibody mutual recognition, the development of novel vaccines, and the development of drugs. Antigen epitopes are a very important aspect in the development of new protein formulations, locating a promising antibody epitope can lead to significant commercial results, and epitope information is critical for patent protection.
Epitope Mapping Services by HDX-MS Workflow
At Creative Proteomics, we offer state-of-the-art epitope mapping services utilizing HDX-MS to elucidate protein-protein interactions and identify binding sites with high precision. Our HDX-MS workflow is designed to provide comprehensive insights into the dynamics of molecular interactions, making it an invaluable tool for researchers in the fields of biochemistry, immunology, and drug development.
Our HDX-MS epitope mapping services includes the following key steps:
- Labeling: Protein samples are incubated with deuterium oxide (D2O) to initiate hydrogen-deuterium exchange.
- Quenching: To halt the exchange reaction, the sample is rapidly cooled and the pH is reduced.
- Digestion: The quenched protein is enzymatically digested into smaller peptides. Proteases such as pepsin or trypsin are used to break down the protein, generating fragments that can be mapped to specific regions of the original protein sequence.
- LC-MS/MS Detection and Data Analysis: The digested peptides are separated using liquid chromatography (LC) and introduced into a mass spectrometer. Tandem mass spectrometry (MS/MS) is employed to measure the mass-to-charge (m/z) ratios of the peptides, providing data on deuterium uptake across different regions of the protein.
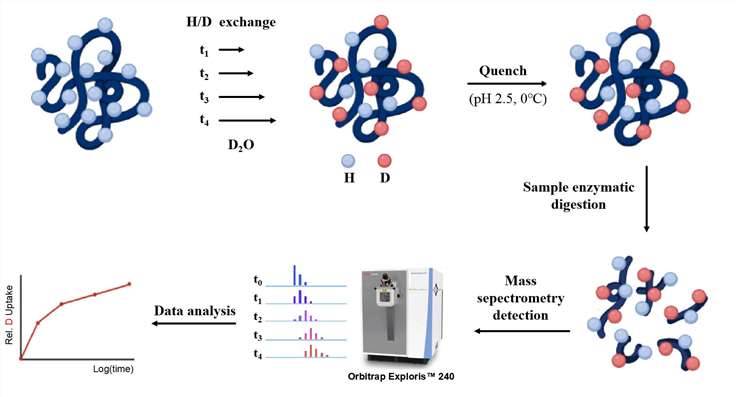
Our Epitope Mapping Service by HDX-MS
Creative proteomics provides state-of-the-art instrumentation and services for HDX-MS technology to rapidly characterize binding sites by HDX-MS. Our epitope mapping service by HDX allows linear and conformational epitope characterization with high resolution and highest success rate. To date, we have successfully identified the epitopes of dozens of antigens with hundreds of antibodies.
Key Features
- 100% successful rate
- Amino acid resolution
- Linear and conformational epitopes
- High-throughput screening of >50 candidates against one antigen
- Accurate mapping for glycosylated antigens by FineMapping technology
- Epitope mapping for unpurified/difficult targets
- Epitope mapping for bispecific antibodies
- Tailored service by top-down, bottom-up, or middle-down HDX-MS
- Unparalleled sensitivity using subzero HPLC technology
- Comprehensive and easy-to-understand report
Other Services that Creative Proteomics HDX-MS provide
- Biosimilar characterization
- Higher-order structure
- Conformational/Protein dynamics
- Small molecules interaction (Compound binding analysis)
- Protein-Protein interactions
- Protein folding characterization
Sample Requirements
- Sample Types: Protein or antibody samples
- Sample Forms: Liquid, dry powder
- Purity: Aim for high-purity protein samples to minimize interference from contaminants.
- Concentration: Ensure that protein concentrations meet our minimum requirements, typically around 1-2 mg/mL for optimal labeling and analysis.
- Buffer Compatibility: Use buffers that do not contain reactive groups or heavy atoms, as these can interfere with deuterium exchange.
- Sample Stability: Confirm that the protein is stable under the experimental conditions; unstable proteins may degrade or denature during the process.
FAQ
Q: What types of proteins or samples can be analyzed with your HDX-MS epitope mapping service?
A: Our epitope mapping service is versatile and can accommodate a wide range of samples, including:
Purified Proteins: High-purity proteins from various sources, including recombinant proteins, can be analyzed.
Complex Protein Mixtures: HDX-MS can also handle samples that consist of multiple interacting proteins, making it suitable for studying protein complexes.
Glycosylated Proteins: The service includes specialized techniques for mapping glycosylated antigens.
Bispecific Antibodies: Our workflow is tailored to study bispecific antibodies, allowing for detailed epitope characterization.
Q: What level of customization do you offer in your epitope mapping services?
A: We recognize that different research projects may have unique requirements. Our epitope mapping services can be tailored to meet specific needs, including:
Choice of HDX-MS Approach: We offer flexibility in selecting the appropriate HDX-MS strategy—top-down, bottom-up, or middle-down—based on the nature of the samples and the desired resolution.
Sample Preparation: We can adapt our sample preparation protocols to accommodate different types of proteins, including those that are difficult to purify or stabilize.
Custom Experimental Design: Our experienced team can work with you to design experiments that align with your research objectives, including specific time points for deuterium labeling or varying pH conditions.
Q: Are there any specific storage or handling recommendations for samples prior to submission?
A: Proper storage and handling of samples are crucial for maintaining their integrity before HDX-MS analysis. Recommendations include:
Temperature Control: Store protein samples at -80°C for long-term storage or 4°C for short-term use to prevent degradation.
Avoid Freeze-Thaw Cycles: Minimize freeze-thaw cycles, as these can lead to protein denaturation. Aliquoting samples can help maintain stability.
Buffer Considerations: Ensure that samples are prepared in compatible buffers (e.g., phosphate-buffered saline) that do not contain reactive species or heavy atoms.
Documentation: Provide detailed information about sample concentration, buffer composition, and any relevant handling notes to facilitate processing.
Case Study
Case: Epitope mapping of polyclonal antibodies by hydrogen–deuterium exchange mass spectrometry (HDX-MS)
Background
The study investigates the binding interactions between polyclonal antibodies (pAbs) and factor H binding protein (fHbp), a 27-kDa lipoprotein crucial for vaccine development against Neisseria meningitidis. fHbp is characterized by two antiparallel β-strand domains and serves as a target for immune recognition. Understanding how antibodies bind to fHbp can provide insights into its immunogenicity and inform vaccine design. Previous studies have highlighted specific epitopes on fHbp recognized by monoclonal antibodies (mAbs), but a comprehensive analysis of the binding profiles of polyclonal antibodies and their relative affinities remains lacking. This study employs HDX-MS to characterize the epitopes recognized by the pAb population against fHbp.
Methods
- Sample Preparation: Recombinant fHbp was produced and incubated with pAbs at different antigen-to-antibody (Ag) ratios (1:2, 1:5, 1:10, 1:15) for HDX-MS experiments.
- HDX-MS Analysis: The exchange of deuterium was monitored over time (from 30s to 20 min) to assess the structural dynamics of fHbp in the presence and absence of pAbs. A sequence coverage of 99.2% was achieved, confirming the robustness of the method.
- Data Interpretation: The differences in deuterium uptake (ΔHDX) between fHbp alone and fHbp bound to pAbs were analyzed to determine regions of protection, indicative of antibody binding.
- VADAR Analysis: The fractional accessible surface area (ASA) of amino acids was assessed using VADAR to identify exposed residues and correlate them with HDX data.
- Avidity and Specificity Testing: The study explored the use of chaotropic agents (like urea and ammonium thiocyanate) and salts (like NaCl) to assess binding specificity and limit nonspecific interactions.
Results
- Identification of Epitopes: The study identified multiple regions on fHbp recognized by pAbs, including N-terminal domains covering residues 3–26, 44–72, and 104–120, which exhibited significant protection from HDX upon pAb binding, and C-terminal areas 133–166 and 209–234 showed protection, with significance emerging at higher pAb concentrations (1:10).
- Binding Affinity Insights: The data suggested that certain antibody populations bind with high affinity, as indicated by the significant changes in HDX with increasing pAb concentrations. This was interpreted as reflecting the relative abundance of binding antibodies rather than their affinity.
- Structural Mapping: The combined VADAR and HDX analyses identified four main epitopic regions, confirming their immunological relevance, including both N- and C-terminal domains. Each identified region had an accessible surface area consistent with previous studies.
- Impact of Additives: The study found that the presence of chaotropic agents and salts affected the HDX results differently.
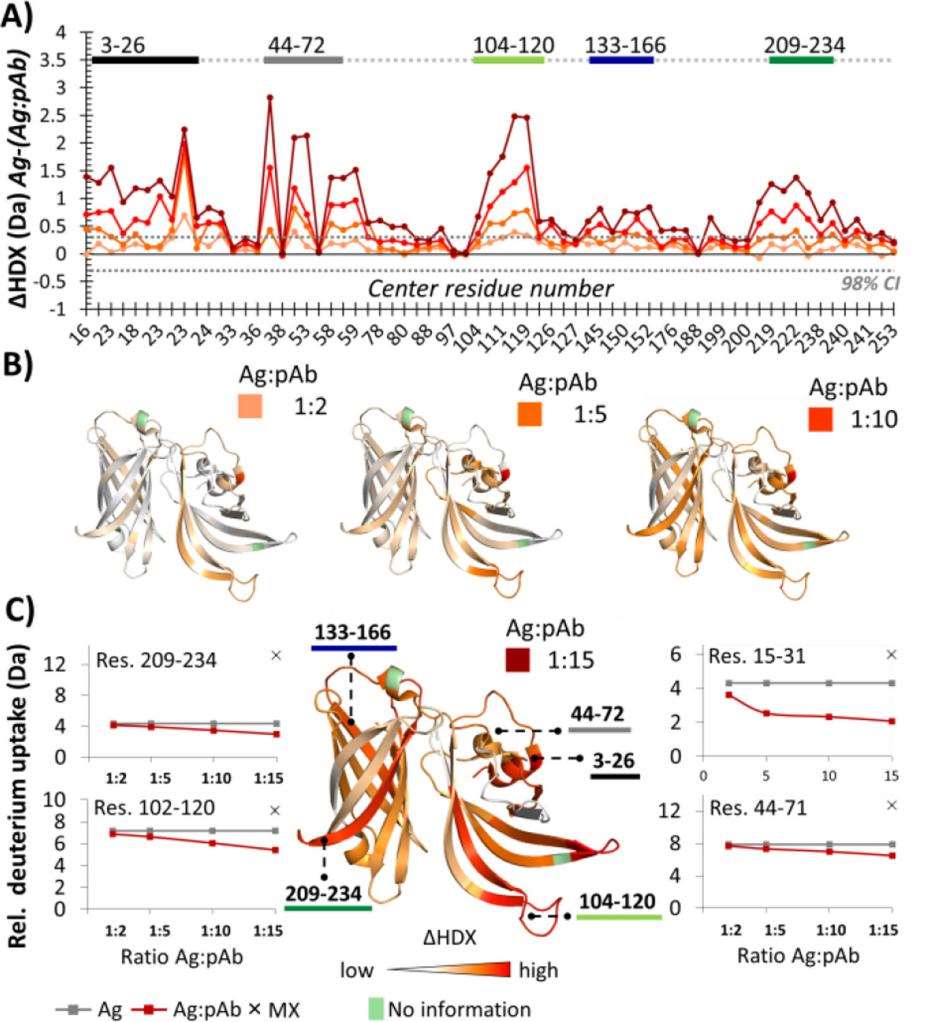 Figure 3. Effect of different Ag:pAb ratios on local HDX of fHbp.
Figure 3. Effect of different Ag:pAb ratios on local HDX of fHbp.References
- Stander S, R. Grauslund L, Scarselli M, et al. Epitope mapping of polyclonal antibodies by hydrogen–deuterium exchange mass spectrometry (HDX-MS). Analytical Chemistry, 2021, 93(34): 11669-11678. DOI: 10.1021/acs.analchem.1c00696
- Puchades C, Kűkrer B, Diefenbach O, et al. Epitope mapping of diverse influenza Hemagglutinin drug candidates using HDX-MS. Scientific reports, 2019, 9(1): 4735. https://doi.org/10.1038/s41598-019-41179-0
- Lim X X, et al. Conformational changes in intact dengue virus reveal serotype-specific expansion. Nature communications, 2017, 8(1): 14339. https://doi.org/10.1038/ncomms14339
- Hudgens J.W., et al., Interlaboratory Comparison of Hydrogen−Deuterium Exchange Mass Spectrometry Measurements of the Fab Fragment of NISTmAb. Analysis chemistry 2019, 91, 7336−7345. DOI: 10.1021/acs.analchem.9b01100
Related Services

Hydrogen Deuterium Exchange Mass Spectrometry Service
Precise, high-quality hydrogen deuterium exchange mass spectrometry service provided by Creative Proteomics to acquire information on protein conformation, dynamics, interactions through drug discovery and development.

Mass Spectrometry based Epitope Mapping Service
Creative Proteomics' epitope mapping service has a success rate of more than 95%, and provides accurate, clear and directly applicable epitope definitions services for antibody projects of drug research and development companies and institutions.

Creative Proteomics employs advanced techniques such as spectroscopy, mass spectrometry, GC/MS, LC/MS, and CE/MS to analyze the structure and stability of your biopharmaceutical formulation.

Creative Proteomics provides highly sensitive, dependable, and precise analysis of tertiary structures, ensuring precise insights into protein conformation and stability for various research applications.
Support Documents



KNOWLEDGE CENTER
The Principle and Application of HDX-MS Protein Structure Analysis

KNOWLEDGE CENTER





