- Services
- FAQ
- Case Study
- Related Services
- Support Documents
- Inquiry
What is Mass Spectrometry Epitope Mapping?
Epitope mapping is a critical process in protein research, focusing on identifying the specific regions, or "epitopes," on an antigen that interact directly with a binding antibody's counterpart, the "paratope." Epitopes are formed by amino acids on the target antigen, and paratopes are formed by amino acids on the binding antibody. Epitopes and paratopes interact to define the location and kinetics of binding. This interaction is the foundation of immune recognition and response.
Epitope mapping is essential in vaccine and drug development. FDA and EMEA guidelines recommend analyzing interaction sites between drugs and targets during regulatory submissions. A thorough understanding of these sites aids in creating more effective, patentable medications.
Creative Proteomics offers an epitope mapping service with a success rate exceeding 95%. This service delivers precise, clear, and directly useful epitope definitions for antibody projects in drug research and development.
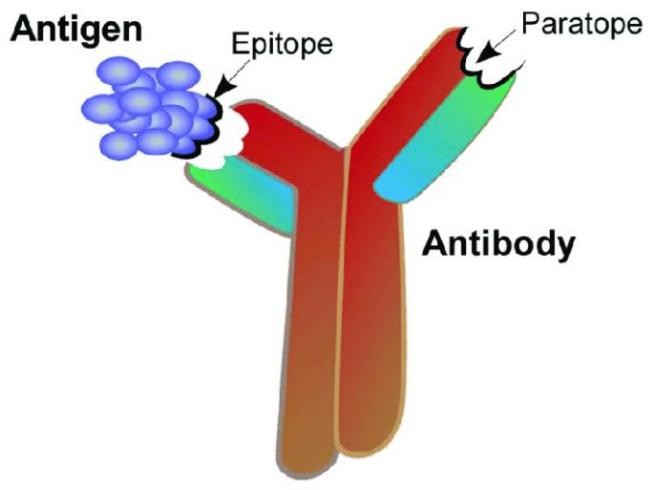
How is Mass Spectrometry Epitope Mapping Used in Protein Analysis?
In protein analysis, mass spectrometry-based epitope mapping provides a comprehensive examination of protein structure, modifications, and interactions. By analyzing peptide fragments resulting from controlled digestion, this technique pinpoints specific protein regions recognized by antibodies, facilitating high-resolution structural analysis and functional characterization of proteins.
A critical aspect of mass spectrometry-based epitope mapping is peptide mapping, which supports the following key analyses:
Verification of Primary Structure: Peptide mapping confirms the amino acid sequence of antibodies and antibody-derived therapeutics, ensuring the accuracy and consistency of the primary structure of protein.
Identification of Point Mutations and Sequence Variants: Epitope mapping enables the detection of point mutations and sequence variants across samples, which is crucial for assessing molecular integrity, especially in biotherapeutics.
Hydrogen-deuterium Exchange Mass Spectrometry (HDX-MS)
HDX-MS measures the accessibility of hydrogen molecules in the protein backbone. During the assay, both unbound antigen and bound antibody-antigen complexes are incubated in deuterium (D2O) water to exchange any amide hydrogens with deuterium from exposed amino acids of the protein backbone. The positions of these deuterium molecules on the protein sequence can then be carefully determined using high-resolution mass spectrometry. By comparing unbound antigen to bound antibody-antigen complexes, the residues of the epitope can be determined. The technique requires careful control of the temperature, pH and time of the reaction.
Utilizing high-resolution molecular structures through HDX analysis can also be used for the following projects:
- Biosimilar Characterization
- Higher order structure
- Conformation/Protein Dynamics
- Small molecule interaction
- Protein-protein interaction
- Protein folding characterization
Cross-Linking Mass Spectrometry (XL-MS)
The ultra-large mass spectrometer first binds antibodies and antigens with mass-tagged chemical cross-linkers. Next, confirm the presence of the complex using high-quality MALDI detection. Since the Ab/Ag complex is very stable after application of cross-linking chemistry, multiple enzymes (five used in parallel) can be applied to the complex to provide many different overlapping peptides. Identification of these peptides was performed using high-resolution Orbitrap™ mass spectrometry and MS/MS techniques. Use a mass tag attached to the crosslinking reagent to determine the identity of the crosslinked peptide. After MS/MS fragmentation and data analysis using specific interaction software, epitopes and paratopes were determined in the same experiment. Due to the high sensitivity and accuracy of mass spectrometric detection, very small amounts of material are required.
Other applications of XL-MS:
- Protein-protein interaction
- Map aggregation
- Paratope mapping analysis
Other Technologies
X-ray co-crystallization
X-ray co-crystallization is often considered the "gold standard" for epitope mapping. If successful, the technique could provide high-confidence resolution of single amino acids. However, this technique is not always feasible, as generating crystals from protein complexes is often difficult and in many cases impossible. This uncertainty often results in long project times. In addition, X-ray analysis requires large protein samples and is costly.
Peptide Scanning
In this technique, a series of overlapping peptides are generated from the antigen. These peptides are then analyzed to see if they disrupt the formation of the complex between the antibody and antigen. This is one of the most common techniques due to its relatively low cost and ability to quickly analyze large numbers of antibodies. This technique is mainly used for the analysis of linear epitopes.
| Item | Linear | Conformational | Complex Protein | High-resolution | Sample Consumption |
|---|---|---|---|---|---|
| HDX-MS | ✔ | ✔ | 450 µg | ||
| XL-MS | ✔ | ✔ | 150ug | ||
| X-ray co-crystallography | ✔ | ✔ | ✔ | tens of mg | |
| Peptide scanning | ✔ | ✔ | tens of µg |
Advantages of Our Service
- Rapid and stable detection of macromolecules and intact protein complexes by MALDI mass spectrometry
- Ensure data accuracy with comprehensive pre-mapping screenings for mass confirmation and multimerization.
- Rapid screening saves our customers time and money.
FAQ
Q: What sample requirements are necessary for HDX-MS and XL-MS analyses?
A: For optimal results in HDX-MS and XL-MS analyses, sample preparation is key, and Creative Proteomics provides guidelines to meet specific method requirements:
HDX-MS: Requires antigens and antibody-antigen complexes at high purity, ideally in D₂O-compatible buffer systems. Concentrations around 0.1–1 mg/mL are typically required to ensure measurable deuterium exchange rates.
XL-MS: Typically requires antibody-antigen samples that are chemically crosslinkable; thus, certain buffer systems are avoided to prevent crosslinking interference. Sample concentration varies depending on target complexity, but typically ranges from 0.1–0.5 mg/mL for accurate crosslinking and analysis.
Q: Can you identify both linear and conformational epitopes using your service?
A: Yes, we can identify both linear and conformational epitopes using HDX-MS and XL-MS:
Linear Epitopes: These consist of contiguous amino acids, and HDX-MS can precisely identify these sites. Additionally, peptide scanning is effective for linear epitope identification.
Conformational Epitopes: These includes amino acids that are spatially close but not necessarily adjacent in the protein sequence. XL-MS is particularly adept at identifying these epitopes, as it can capture cross-links between distant regions of the protein that come together in the folded state.
Case Study
Case: Epitope mapping on bovine prion protein using chemical cross-linking and mass spectrometry
Background
This study presents a rapid and reliable approach to epitope mapping, crucial for developing therapeutic and diagnostic antibodies. It addresses the limitations of traditional methods like PEPSCAN, which are time and resource-intensive. The limitations of these conventional techniques underscore the importance of developing more efficient strategies to accelerate epitope identification and improve the overall process of antibody development. The study explores an alternative approach using chemical cross-linking combined with high-mass MALDI-TOF MS and nano-LC-ESI-FTICR MS. This combined method is proposed as a faster and more efficient strategy for identifying epitopes, even in complex antigen-antibody systems.
Methods
The researchers employed cross-linkers DSS and DSG, reagents typically targeting primary amines like those in lysine residues, to investigate cross-linking reactivity with the antigen-antibody complex. The experiment involved several key steps:
- Chemical Cross-Linking: DSS and DSG were applied to cross-link the antigen bPrP(25–241) with the monoclonal antibody 3E7. The conditions were optimized for reactivity, with considerations given to molar excess and reaction time.
- Mass Spectrometry Analysis: Cross-linked peptides were analyzed using high-mass MALDI-TOF MS to assess the intact antigen-antibody complexes and determine their stoichiometry. High-resolution nano-LC-ESI-FTICR MS was also used to identify specific peptides involved in cross-linking.
- MS/MS Analysis: Selected peptides were further analyzed using tandem MS (MS/MS) to identify exact modification sites, providing insight into the side-chain reactivity of DSS and DSG.
Results
The results highlighted several key findings:
- Unexpected Side-Chain Reactivity: While DSS and DSG are designed to target primary amines on lysine residues, the study found significant reactivity with hydroxyl groups on tyrosine and serine residues as well. This was confirmed in peptide [150–160], where modifications were observed on tyrosine and serine, despite the absence of lysine. This finding aligns with previous studies and suggests that N-hydroxysuccinimide esters can react with side chains beyond primary amines.
- Epitope Mapping Success: The cross-linking strategy successfully identified the epitope region on bPrP(25–241) recognized by the monoclonal antibody 3E7. The identified peptides were consistent with PEPSCAN data, demonstrating the reliability of this approach in mapping linear epitopes.
- Improved Efficiency and Sensitivity: The combined method of chemical cross-linking and MS analysis proved to be efficient, requiring only picomole quantities of protein. Additionally, the method demonstrated high sensitivity in detecting cross-linking products, particularly when high-resolution FTICR-MS instrumentation was used.
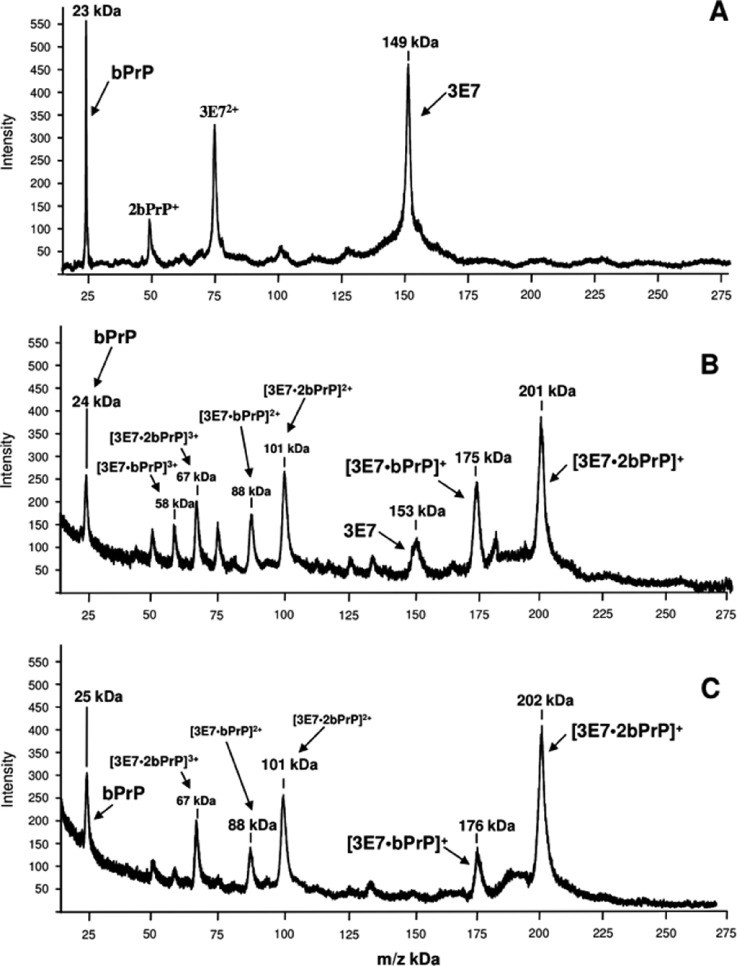 Figure 1. High-mass MALDI-TOF mass spectra of the immuno complexes formed between bPrP and the monoclonal antibody 3E7.
Figure 1. High-mass MALDI-TOF mass spectra of the immuno complexes formed between bPrP and the monoclonal antibody 3E7. Figure 2. Peptide mass fingerprint of bPrP(25–241) obtained after chymotryptic proteolysis.
Figure 2. Peptide mass fingerprint of bPrP(25–241) obtained after chymotryptic proteolysis.![FTICR mass spectra of the peptides [150–160] and [230–237].](upload/image/mass-spectrometry-based-epitope-mapping-service-figure-4.jpg) Figure 3. FTICR mass spectra of the peptides [150–160] and [230–237] (triply charged signals) analyzed after chymotryptic digestion of the bovine prion protein following different treatments.
Figure 3. FTICR mass spectra of the peptides [150–160] and [230–237] (triply charged signals) analyzed after chymotryptic digestion of the bovine prion protein following different treatments.![MS/MS analysis of the peptide [150–160] modified with DSS-d0 and DSS-d12.](upload/image/mass-spectrometry-based-epitope-mapping-service-figure-5.jpg) Figure 4. MS/MS analysis of the peptide [150–160] modified with (A) DSS-d0 and (B) DSS-d12. The sequence of the peptide is shown with masses for the b and y fragments.
Figure 4. MS/MS analysis of the peptide [150–160] modified with (A) DSS-d0 and (B) DSS-d12. The sequence of the peptide is shown with masses for the b and y fragments.References
- Opuni K F M, et al. Mass spectrometric epitope mapping. Mass spectrometry reviews, 2018, 37(2): 229-241. DOI: 10.1002/mas.21516
- Liu X R, et al. Advances in mass spectrometry-based epitope mapping of protein therapeutics. Journal of pharmaceutical and biomedical analysis, 2022, 215: 114754. DOI: 10.1016/j.jpba.2022.114754
- Jethva P N, Gross M L. Hydrogen deuterium exchange and other mass spectrometry-based approaches for epitope mapping. Frontiers in analytical science, 2023, 3: 1118749. DOI: 10.3389/frans.2023.1118749
- Pimenova T, et al. Epitope mapping on bovine prion protein using chemical cross‐linking and mass spectrometry. Journal of mass spectrometry, 2008, 43(2): 185-195. DOI: 10.1002/jms.1280
Related Services

Hydrogen Deuterium Exchange Mass Spectrometry (HDX-MS) Service
Precise, high-quality hydrogen deuterium exchange mass spectrometry service provided by Creative Proteomics to acquire information on protein conformation, dynamics, interactions through drug discovery and development.

Epitope Mapping Services by HDX-MS
Creative proteomics provides state-of-the-art instrumentation and services for HDX-MS technology to rapidly characterize binding sites by HDX-MS.

Creative Proteomics have a strict workflow to analyze X-ray Crystallography to meet your requirements.
Support Documents


KNOWLEDGE CENTER
The Principle and Application of HDX-MS Protein Structure Analysis

KNOWLEDGE CENTER





