- Services
- FAQ
- Demo
- Case Study
- Related Services
- Support Documents
- Inquiry
What is Intact Protein Analysis?
Intact proteins are crucial for biological functions like enzymatic reactions, signaling, and immune responses. Intact mass analysis measures their molecular weight in their complete form, key for characterizing biotherapeutics like monoclonal antibodies. This technique reveals protein structure, heterogeneity, and post-translational modifications (PTMs). The best methods for intact mass analysis are MALDI-MS and ESI-MS, offering insights into protein composition and function, essential for developing and controlling therapeutic proteins.
The significance of intact mass analysis extends beyond simple molecular weight determination. It helps identify variants and degradation products, providing detailed characterization crucial for the efficacy and safety of protein therapeutics in the biopharmaceutical industry.
From early research and development, characterization analysis, process optimization to CMC and QC analysis, biopharmaceuticals have complexity and uncertainty in every link. Creative Proteomics provides high resolution mass spectrometry biopharmaceutical solutions to support your product from development to production.
The Principle of ESI-MS
ESI-MS is a special soft ionization technology that can generate gas phase ions with multiple charges more gently, reducing the mass-to-charge ratio range of the molecules to be measured. ESI-MS capitalizes on the ability to ionize large biomolecules without fragmentation. In ESI-MS, a protein solution is nebulized into a fine aerosol of charged droplets. As these droplets evaporate, ions are produced, allowing for the analysis of the intact protein.
ESI-MS measures the mass-to-charge (m/z) ratio of ions from protein molecules. The data can be deconvoluted to provide accurate molecular weight values, offering crucial information on protein structure and modifications.
ESI-MS not only can obtain accurate molecular weight of compounds, but also can be used for quantitative and qualitative analysis of trace or trace biologically active molecules. It is also suitable for studying weak bond interactions. Combined with collision-induced dissociation technology, electrospray ionization mass spectrometry can also be used for structural analysis, reaction mechanism, and non-covalent bonding capabilities.
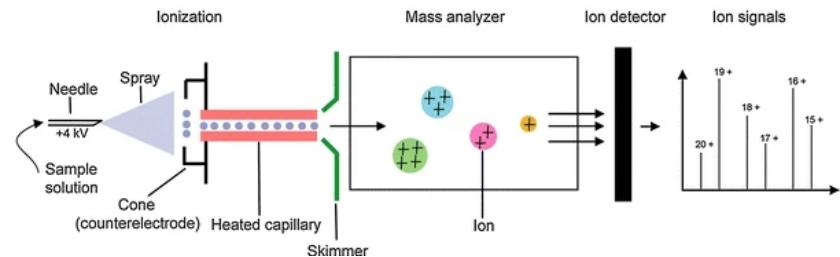 Figure 1. The procedure of ESI-MS (Lin Y, et al., 2009)
Figure 1. The procedure of ESI-MS (Lin Y, et al., 2009)We Provide but Not Limited to
- Molecular weight determination
- Peptide map analysis
- Analysis of biopharmaceutical host cell residual proteins (HCPs)
- Study the folding path of proteins: analyze the conformational changes of protein molecular ions, obtain the transition state information of conformational changes, and understand the process of spatial conformation.
- Study the non-covalent effect of peptides and metals
- Study the non-covalent effect of protein and protein, peptide and peptide, protein and DNA, protein and peptide: intramolecular binding force or non-covalent binding selectivity, affinity, stability, determination of binding site, calculation of binding constant or dissociation constant.
Why Choose Our ESI-MS Intact Protein Analysis Service
- High sensitivity, high accuracy, easy to operate and fast detection.
- ESI-MS can be effectively used in combination with chromatography, and is suitable for the identification or structure determination of trace substances in complex systems.
- Suitable for the analysis of strong polarity, difficult volatilization or thermal instability compounds.
- The limited detection conditions are few and the signal is easy to be analyzed.
- The information of transition state can be obtained, and the composition and conformation of polypeptide and protein molecules can be studied effectively.
Sample Requirements
- Purity>90%
- Avoid detergents and keep buffer concentration at a minimum
- Quantity>40ug
- Acceptable buffers: ammonium acetate, ammonium bicarbonate, ammonium formate.
- Avoiding buffer: HEPES, PBS, MES, MOPS, and Tris.
- Avoiding detergents: SDS, PEG, PPG, Tween, CHAPS, Triton, and urea.
- Avoiding salts: Alkylammonium salts, guanidinium salt, metal cations and inorganic anions such as phosphate, sulfate, and halides.
Creative Proteomics' analytical scientists can provide customers with ESI-MS intact protein analysis service. We will deliver raw data, concise and clear results report, detailed bioinformatics analysis reports.
FAQ
Q: Can ESI-MS analyze non-covalent interactions between proteins and other molecules?
A: Yes, ESI-MS can analyze non-covalent interactions, such as protein-protein, protein-ligand, and protein-DNA bindings, by preserving weak bonds during ionization. It helps study binding affinity, stability, and dissociation constants, crucial for understanding drug development and protein function.
Q: How does ESI-MS differ from MALDI-TOF in intact protein analysis, and when is one preferred over the other?
A: Both ESI-MS and MALDI-TOF-MS are widely used for intact protein analysis, but they differ in their ionization techniques and best-suited applications:
ESI-MS: ESI-MS is ideal for large, fragile biomolecules, preserving non-covalent interactions through soft ionization. It generates multiply charged ions, enabling analysis of large proteins and complexes.
MALDI-TOF-MS: This method generates singly charged ions and is often faster, but less suited for complex proteins or those with weak bonds that may be disrupted during ionization. MALDI is commonly used when a rapid molecular weight measurement is needed for smaller or more stable proteins. Each method has its strengths, you can learn more details in the article: Comparison of MALDI and ESI.
Q: Does ESI-MS work for membrane proteins?
A: Yes, ESI-MS can be used for membrane proteins, but challenges arise due to their hydrophobic nature and the need for solubilizing agents. Common detergents like SDS and Triton interfere with ionization, so alternative strategies such as mild non-ionic detergents or amphipathic polymers are required. Creative Proteomics offers guidance for such challenging proteins.
Q: How does ESI-MS handle proteins with multiple subunits or quaternary structures?
A: ESI-MS analyzes protein complexes and their intact molecular weight, including quaternary structures or multi-subunit assemblies. By preserving non-covalent interactions, it detects protein-protein interactions. However, as subunits increase, mass spectrum complexity grows, making deconvolution harder. CID can be used for subunit analysis.
Demo
Demo 1: ESI-MS Spectra of IGF-1, Recombinant Human Growth Hormone, the F(ab')2 Part of antibody and antibody.
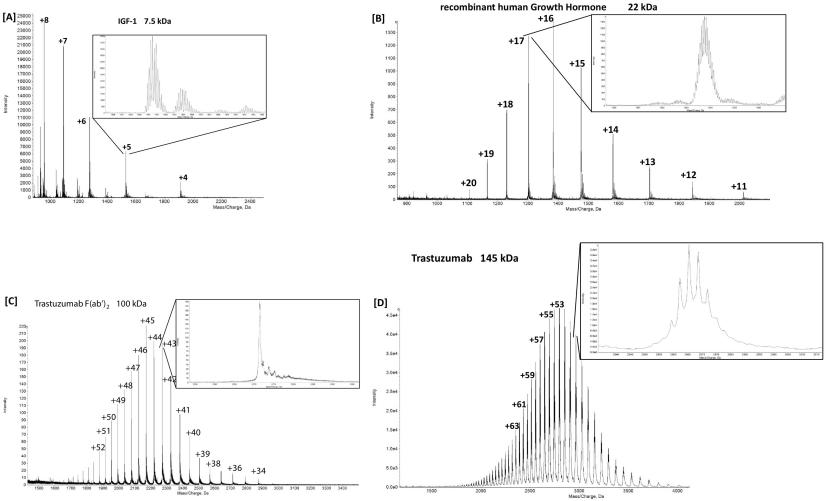 Figure 2. Charge state envelope of IGF-1 (A), recombinant human growth hormone (B), the F(ab′)2 part of antibody (C) and antibody (D) with a zoom into one charge state.
Figure 2. Charge state envelope of IGF-1 (A), recombinant human growth hormone (B), the F(ab′)2 part of antibody (C) and antibody (D) with a zoom into one charge state.References
- Bults P, et al. Intact protein bioanalysis by liquid chromatography–High-resolution mass spectrometry. Journal of Chromatography B, 2019, 1110: 155-167.
Case Study
Case: Quantification of intact covalently metal labeled proteins using ESI-MS/MS
Background
The article presents a novel approach for protein quantification using lanthanide-based labels in conjunction with mass spectrometry (MS). The study focuses on the application of MeCAT-IA (metal-coded affinity tag) labeling for intact proteins, which binds to cysteine residues, allowing for simultaneous identification and quantification. The method is especially relevant for large, complex proteins like human serum albumin (HSA). Traditional techniques, such as isotope dilution and ICP-MS (inductively coupled plasma mass spectrometry), are established for absolute quantification, but they involve digestion of proteins, which can lead to losses and inconsistencies. The authors aim to demonstrate the efficacy of MeCAT-IA labeling combined with electron spray ionization (ESI)-MS/MS for direct quantification of intact proteins, circumventing the need for digestion.
Methods
- Labeling: Proteins were labeled with MeCAT-IA, which specifically binds to cysteine residues. For the study, five lanthanide isotopes (Eu, Tb, Ho, Tm, Lu) were used, enabling multiplex quantification.
- Mass Spectrometry: Labeled proteins were analyzed using infrared multiphoton dissociation (IRMPD) and higher-energy collisional dissociation (HCD) fragmentation with Orbitrap-based MS/MS for mass spectrometry.
- Quantification: The authors evaluated the labeled proteins by comparing the fragmentation patterns and intensities, particularly focusing on the cysteine-containing fragments. They validated the data by comparing the results with isotope dilution-based ICP-MS measurements, a gold standard for quantitative analysis. The quantification of HSA in human blood serum served as a proof-of-concept for the method's applicability to real biological samples.
- Instrument Limitations: They also assessed the limitations of the Q Exactive mass spectrometer, particularly the 10 u isolation window, which caused challenges in isolating precursor ions in complex protein mixtures.
Results
- Consistency with ICP-MS: The IRMPD and HCD fragmentations produced quantifiable ion signals that were consistent with the results from ICP-MS. This confirms that MeCAT-IA labeling with MS/MS analysis provides accurate quantification, validating the approach for both peptides and intact proteins.
- HSA Quantification: The method was applied to human blood serum HSA, with concentrations measured at 51.6 ± 5.8 mg/mL using the new MS/MS method, compared to 53.4 ± 3.9 mg/mL obtained by ICP-MS. Both values were in good agreement, reinforcing the method's precision.
- Multiplexing Capability: The MeCAT-IA technique allowed for the simultaneous analysis of five different protein samples, demonstrating the method's potential for multiplex quantification in complex biological samples.
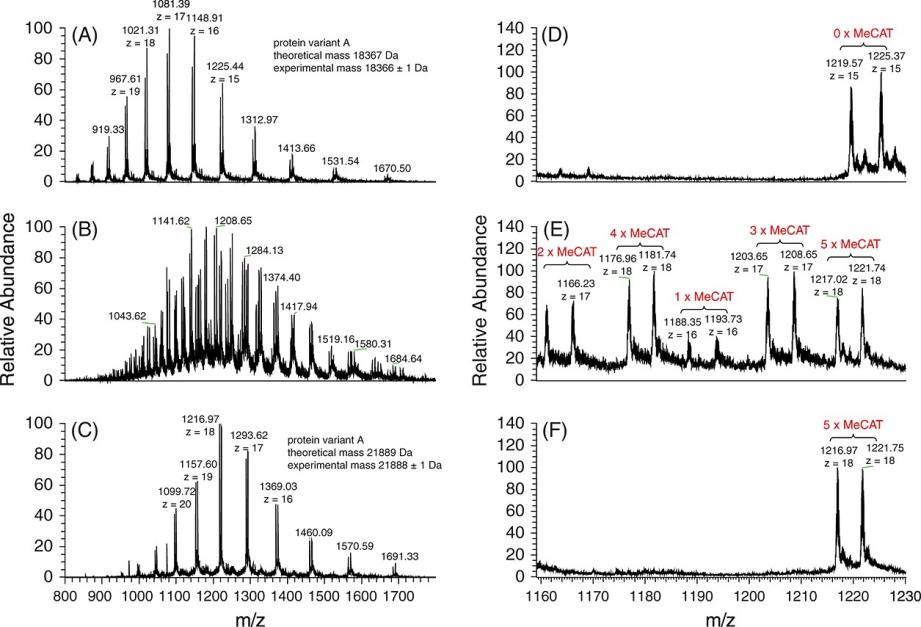 Figure 3. ESI-FTICR-MS spectra of beta-lactoglobulin.
Figure 3. ESI-FTICR-MS spectra of beta-lactoglobulin.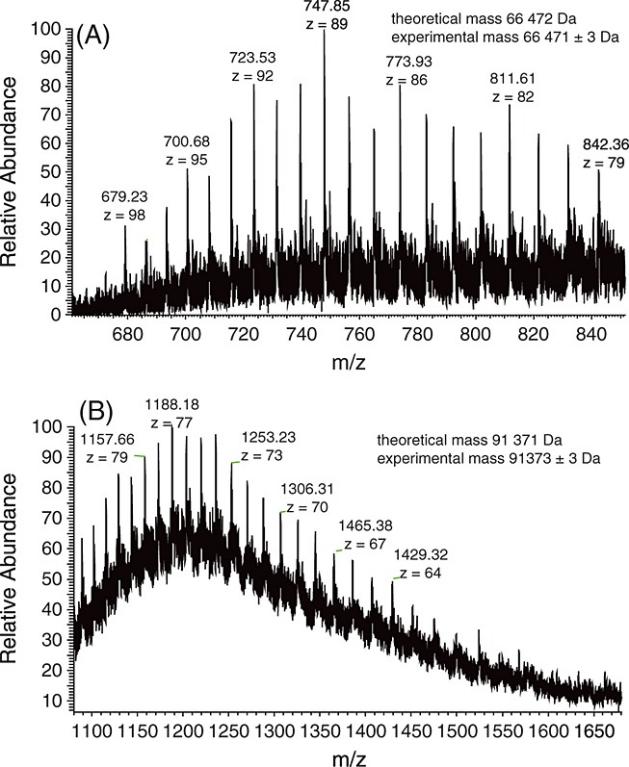 Figure 4. ESI-FTICR-MS spectra of (a) reduced HSA and (b) HSA labeled with Ho-MeCAT-IA.
Figure 4. ESI-FTICR-MS spectra of (a) reduced HSA and (b) HSA labeled with Ho-MeCAT-IA.References
- Benda D, et al. Quantification of intact covalently metal labeled proteins using ESI‐MS/MS. Journal of Mass Spectrometry, 2014, 49(1): 13-18. DOI: 10.1002/jms.3302
- Lin Y, et al. The current state of proteomics in GI oncology. Digestive diseases and sciences, 2009, 54: 431-457. doi: 10.1007/s10620-008-0656-5
- Loo J A. Electrospray ionization mass spectrometry: a technology for studying noncovalent macromolecular complexes. International Journal of Mass Spectrometry, 2000, 200(1-3): 175-186. https://doi.org/10.1016/S1387-3806(00)00298-0
Related Services

Intact Mass Analysis Service of Therapeutic Proteins
Discover the role of intact mass analysis in protein characterization for drug development and Creative Proteomics' protein analysis services.

Protein Molecular Weight Determination Service
Creative Proteomics is equipped with both of MALDI-TOF-MS and ESI-MS mass spectrometry systems which can provide customers with accurate determination of protein molecular weight.

MALDI-TOF-MS Intact Protein Analysis
Elevate your biopharmaceutical quality assessment with MALDI-TOF-MS intact protein analysis. Accurate mass measurement for proteins and peptides from 1 to 100 kDa. Contact us for high-resolution analysis

LC-MS Analysis of Intact Antibodies
For intact antibody analysis, Creative Proteomics has developed a highly sensitive and stable LC/MS workflow. Our workflow is based on optimized sample preparation, improved LC-MS instrumentation, and advanced analytical software.
Support Documents










