- Services
- FAQ
- Demo
- Case Study
- Related Services
- Support Documents
- Inquiry
What is Hydrogen Deuterium Exchange Mass Spectrometry (HDX-MS)?
Hydrogen deuterium exchange mass spectrometry (HDX-MS) is utilized to investigate protein conformation and dynamics, as well as interactions among proteins, small molecules, and RNA. It leverages the inherent proton exchange that occurs at the amide bonds within proteins. When proteins are exposed to a deuterium oxide (D₂O)-based buffer, the hydrogens in the protein backbone can be substituted with deuterium. According to the protein's structure, the exchange rates varies. If the hydrogen-bonded segments are tight, the exchange rate is low; the exchange rate is high when the regions are disordered. The hydrogens on the protein surface will be exchanged with deuterium atoms after immersing the protein or protein complex into the deuterium solution. The exchange will be then detected and quantified by mass spectrometry since the deuterium is heavier than hydrogen. HDX-MS can be utilized to detect single protein's and protein complexes' structure and conformation information, as well as protein-protein or protein-ligand interaction sites and conformational changes induced by post-translational modifications (PTMs) in protein complexes.
Since the high order structure and conformational dynamics of proteins are closely related to the efficacy of biopharmaceutical agents, it is essential to well acknowledge protein advanced structure, conformation, and interactions during drug discovery and development. Creative Proteomics provides a precisive HDX-MS service to discover information on protein conformation, dynamics, interaction sites in various types of samples, including monoclonal antibodies (mAbs), antibody-drug conjugates (ADCs), viral capsids, and so on. Our professional scientists will optimize procedures to fulfill the requirements during all stages of drug discovery and development.
 Figure 1. Hydrogen deuterium exchange mass spectrometry in biopharmaceutical discovery and development (Bin Deng et al., 2016)
Figure 1. Hydrogen deuterium exchange mass spectrometry in biopharmaceutical discovery and development (Bin Deng et al., 2016)How HDX-MS Works?
The HDX-MS process involves several coordinated steps designed to capture the dynamic behavior of proteins in solution:
- Preparation: Proteins of interest are prepared (or produced in the lab) and placed in a deuterated solvent (to exchange hydrogens in the protein for deuterium atoms).
- Quenching the Exchange Reaction: To stop the exchange and prevent undesirable back-exchange of deuterium to hydrogen the reaction process is quenched by a low pH buffer.
- Proteolytic Digestion: The protein samples are quenched with acid, and then proteolyzed by specific enzymes (for instance, pepsin, trypsin).
- Mass Spectrometric Analysis: Peptides are examined using mass spectrometry (MS). The peptides are ionized, and their mass-to-charge ratios are measured.
- Data Interpretation: The data on deuterium uptake is then analyzed to pinpoint areas of the protein that undergo structural changes, particularly in response to binding events or varying experimental conditions.
HDX-MS Technology Platform
In HDX experiments, protein will be first labeled. Two main approaches can be performed to label proteins by HDX: continuous labeling and pulsed labeling. In continuous labeling approach, several aliquots of the protein are immersed in the deuterated buffer under conditions that keep the native conformation of the protein. Each aliquot of the protein sample is incubated in the buffer for a certain period time and then labeling is ended. This approach can monitor the deuterium exchange as a function of time, and be used to investigate conformational dynamics of a protein under equilibrium conditions. In pulsed labeling approach, a protein is first perturbed by chemical denaturant, pH change, temperature change, etc. Then the protein is immersed in deuterated buffer with higher pH for a short time. This approach allows HDX occur only at positions that are solvent-exposed but not involved in hydrogen bonds. In order to reduce the exchange rate and make an end to the reaction during analysis, the deuterated protein will be placed in a low pH buffer at low temperature following labeling. Subsequently, MS will be performed to measure the deuterium incorporation into the proteins.
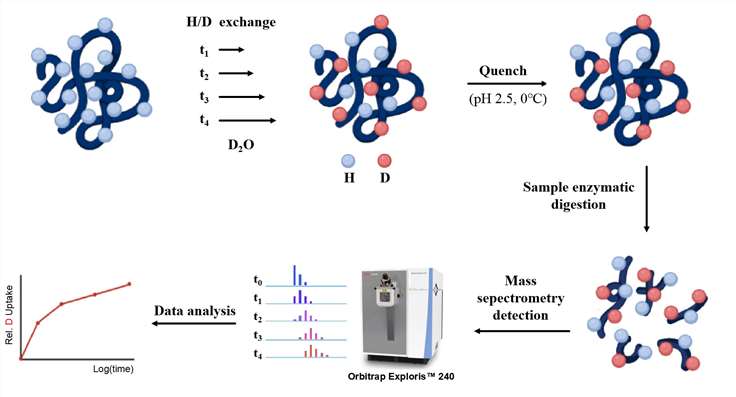
We Provide but Not Limited to
- Identification of protein conformation
- Epitope mapping
- Determination of protein-protein interaction, protein-small molecule interactions, and protein-RNA interactions
- Assessments of dynamical comparability
- Evaluation of structural modification
Why Choose Our ESI-MS Intact Protein Analysis Service
- High sensitivity and accuracy: precise characterization of proteins with different molecular weight from ~10 kDa to ~2MDa.
- Good batch to batch reproducibility.
- Advanced system: Orbitrap Exploris™ 240 Mass Spectrometer, etc.
- Customized service: Optimized methods will be customized based on your sample type, size and service.
Creative Proteomics's experienced scientists can provide extensive analysis of protein by HDX-MS, including conformation, interaction, etc. Clear, comprehensive written reports, recommendations and protocols, and customized services will be provided to help customers solve analytical and technical problems.
Sample Requirements
To ensure successful HDX-MS analysis, we recommend the following guidelines for sample preparation:
- Protein Concentration: 1 to 10 mg/mL (lower concentrations may be acceptable).
- Sample Volume: At least 50–100 µL.
- Buffer Conditions: Samples should be in HDX-compatible buffers (e.g., phosphate or HEPES) and free of detergents, salts, or interfering substances.
- Purity: Proteins should be highly pure (≥95%), with minimal contaminants or aggregation.
- Stability: Ensure protein stability under the conditions used, avoiding factors that may cause degradation or aggregation.
FAQ
Q: How does HDX-MS complement other structural biology techniques?
A: Traditional structural biology techniques such as X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy offer static, high-resolution representations of protein structures, HDX-MS provides dynamic insights by monitoring protein conformational changes and flexibility in solution. This capacity for detecting transient, flexible, or disordered regions of proteins, which may be elusive or unresolved in other techniques, renders HDX-MS a highly complementary tool. It proves particularly valuable in elucidating regions of structural plasticity that are critical for protein function but may remain undetected through conventional static approaches.
Q: How does HDX-MS handle complex protein mixtures?
A: HDX-MS demonstrates significant flexibility in analyzing complex protein mixtures, although increased sample complexity can introduce challenges in data interpretation. This approach enables the precise quantification of deuterium incorporation at specific regions within individual proteins, even in the context of a complex matrix. Furthermore, high-performance liquid chromatography (HPLC) is often incorporated to enhance peptide separation prior to mass spectrometric analysis, thereby improving the resolution and interpretability of HDX-MS data. This capability allows for detailed structural insights even in intricate protein assemblies.
Demo
Demo 1: The HDX data of helix 3, the β-sheet region, helix 12 and the helix 2–2′ link region containing the site of CDK5 phosphorylation.
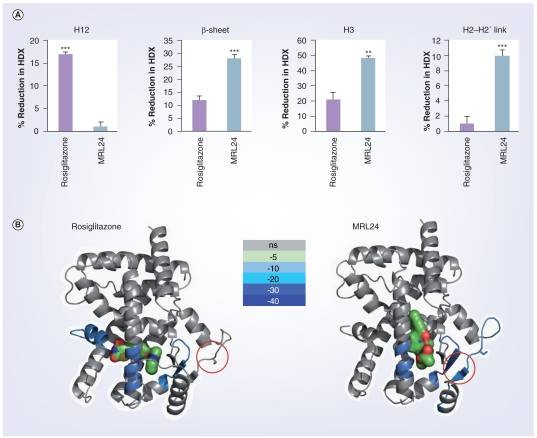 Figure 2. The HDX data shown correspond to four regions of interest: helix 3, the β-sheet region , helix 12 and the helix 2-2′ link region containing the site of CDK5 phosphorylation.
Figure 2. The HDX data shown correspond to four regions of interest: helix 3, the β-sheet region , helix 12 and the helix 2-2′ link region containing the site of CDK5 phosphorylation.Case Study
Case: Hydrogen/deuterium exchange mass spectrometry and computational modeling reveal a discontinuous epitope of an antibody/TL1A Interaction
Background
This article focuses on epitope mapping using electron transfer dissociation (ETD) - enhanced hydrogen deuterium exchange mass spectrometry (hdx-ms) to improve the spatial resolution of mapping antibody antigen interactions. This study highlighted the importance of epitope mapping in the development of therapeutic antibodies, especially for IgG4 anti-tl1a monoclonal antibody (MAb1), which effectively blocks tl1a/dr3 interaction. Tl1a is a homotrimeric protein that is crucial in immune signaling, and understanding its binding mechanism is crucial for designing antibodies with improved efficacy. Traditional methods such as X-ray crystallography and nuclear magnetic resonance (NMR) are often hampered by the complexity of protein complexes, which has prompted the need for faster and more effective technologies, such as hdx-ms.
Methods
The research employed a combination of HDX-MS and ETD to refine the spatial resolution of epitope mapping:
- Sample Preparation: The TL1A protein and its complex with mAb1 were expressed in CHO cells and purified. The experiments utilized a synthetic peptide to optimize deuterium scrambling.
- Hydrogen-Deuterium Exchange (HDX): Non-deuterated experiments were performed to identify common peptides for TL1A. The protein was labeled with deuterium in varying time intervals to assess deuterium uptake.
- Mass Spectrometry and Fragmentation: Peptides were fragmented using ETD, which minimizes deuterium scrambling compared to traditional methods. The deuterium uptake was measured for various peptide fragments to assess structural dynamics and binding interactions.
- Computational Analysis: Solvent accessible surface area (SASA) calculations were performed alongside molecular docking to visualize and predict the binding interactions between TL1A and mAb1. The structural models were validated through multiple computational approaches.
Results
- Epitope Identification: Two significant regions on TL1A, residues 102–116 and 166–180, were identified as potential binding epitopes for mAb1 through differential HDX and SASA analysis.
- Binding Mechanism Insights: The study provided insights into the conformational dynamics of TL1A upon mAb1 binding, suggesting that residues 113KNQF116 and 169QAGR172 play key roles in antibody interaction.
- Correlative Data: There was a strong correlation between HDX-ETD results and SASA analysis, with specific residues identified as functional and solvent-exposed, further supporting their role in binding.
- Structural Stability: The HDX kinetics showed that the TL1A subunit interface was solvent-protected and stable, while the outer loop regions were more flexible and accessible.
- Functional Impact: Functional assays demonstrated that mAb1 efficiently inhibited TL1A binding to the DR3 receptor, indicating the practical implications of the identified epitopes in therapeutic applications.
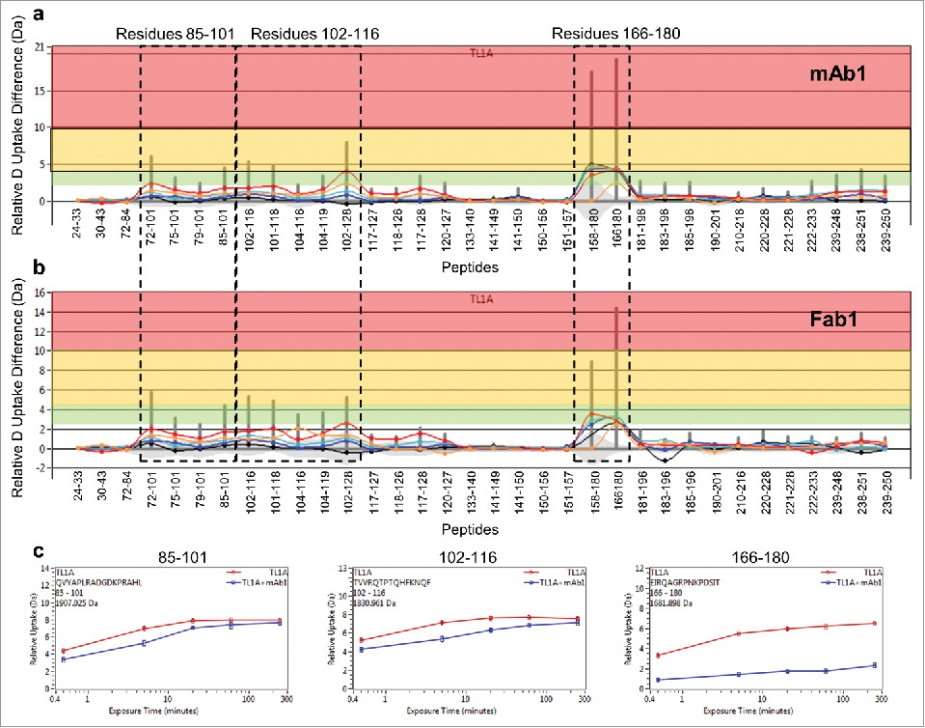 Figure 3. Differential HDX of TL1A upon (a) mAb1 binding and (b) Fab1 binding.(c) HDX kinetics curves of 85QVYAPLRADGDKPRAHL101, 102TVVRQTPTQHFKNQF116 and 166EIRQAGRPNKPDSIT180.
Figure 3. Differential HDX of TL1A upon (a) mAb1 binding and (b) Fab1 binding.(c) HDX kinetics curves of 85QVYAPLRADGDKPRAHL101, 102TVVRQTPTQHFKNQF116 and 166EIRQAGRPNKPDSIT180.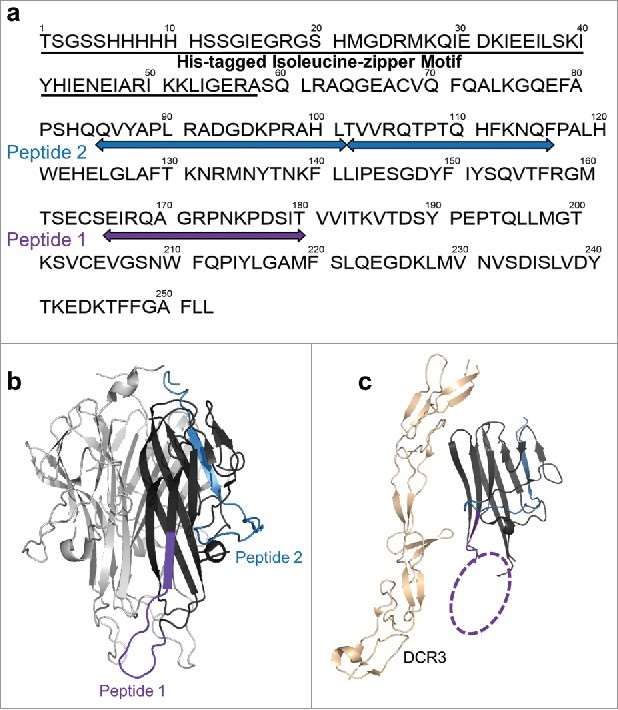 Figure 4. HDX revealed two major epitope regions of mAb1, peptide 1 and peptide 2. (a) Epitope mapped onto the TL1A protein sequence. The TL1A construct is composed of the His-tagged isoleucine-zipper motif followed by the extracellular domain; (b) TL1A trimer crystal structure (PDB code: 2RE9); and (c) TL1A/DcR3 crystal structure (PDB code: 3K51).
Figure 4. HDX revealed two major epitope regions of mAb1, peptide 1 and peptide 2. (a) Epitope mapped onto the TL1A protein sequence. The TL1A construct is composed of the His-tagged isoleucine-zipper motif followed by the extracellular domain; (b) TL1A trimer crystal structure (PDB code: 2RE9); and (c) TL1A/DcR3 crystal structure (PDB code: 3K51).References
- Hudgens J.W., et al., Interlaboratory Comparison of Hydrogen−Deuterium Exchange Mass Spectrometry Measurements of the Fab Fragment of NISTmAb. Anal Chem, 2019, 91, 7336−7345. DOI: 10.1021/acs.analchem.9b01100
- Deng B., Lento C., and Wilson D.J., Hydrogen deuterium exchange mass spectrometry in biopharmaceutical discovery and development-A review. Analytica Chimica Acta, 940 (2016) 8e20. DOI: 10.1016/j.aca.2016.08.006
- Wei H., et al., Hydrogen/Deuterium Exchange Mass Spectrometry for Probing Higher Order Structure of Protein Therapeutics: Methodology and Applications. Drug Discov Today, 2014 January; 19(1): 95–102. DOI: 10.1016/j.drudis.2013.07.019
- Lin Y, et al. The current state of proteomics in GI oncology. Digestive diseases and sciences, 2009, 54: 431-457. DOI: 10.1007/s10620-008-0656-5
- Loo J A. Electrospray ionization mass spectrometry: a technology for studying noncovalent macromolecular complexes. International Journal of Mass Spectrometry, 2000, 200(1-3): 175-186. DOI: 10.1016/s1387-3806(00)00298-0
- Chalmers, et al., Differential hydrogen/deuterium exchange mass spectrometry analysis of protein-ligand interactions. Expert Rev Proteomics, 2011. 8(1): p. 43-59. DOI: 10.1586/epr.10.109
- Huang R Y C, et al. Hydrogen/deuterium exchange mass spectrometry and computational modeling reveal a discontinuous epitope of an antibody/TL1A interaction. MAbs. Taylor & Francis, 2018, 10(1): 95-103. DOI: 10.1080/19420862.2017.1393595
Related Services

Epitope Mapping Services by HDX-MS
Creative proteomics provides state-of-the-art instrumentation and services for HDX-MS technology to rapidly characterize binding sites by HDX-MS.

Creative Proteomics employs advanced techniques such as spectroscopy, mass spectrometry, GC/MS, LC/MS, and CE/MS to analyze the structure and stability of your biopharmaceutical formulation.

Creative Proteomics provides highly sensitive, dependable, and precise analysis of tertiary structures, ensuring precise insights into protein conformation and stability for various research applications.
Support Documents



KNOWLEDGE CENTER
The Principle and Application of HDX-MS Protein Structure Analysis

KNOWLEDGE CENTER





