The genetic stability and accuracy of gene expression systems are paramount for the production of high-quality monoclonal antibody drugs. Comprehensive sequence analysis, encompassing DNA, mRNA, and protein levels, ensures the consistency and safety of biopharmaceutical products. The implementation of advanced sequencing techniques and stability assessments, alongside regulatory compliance, forms the foundation for successful biopharmaceutical development and manufacturing.
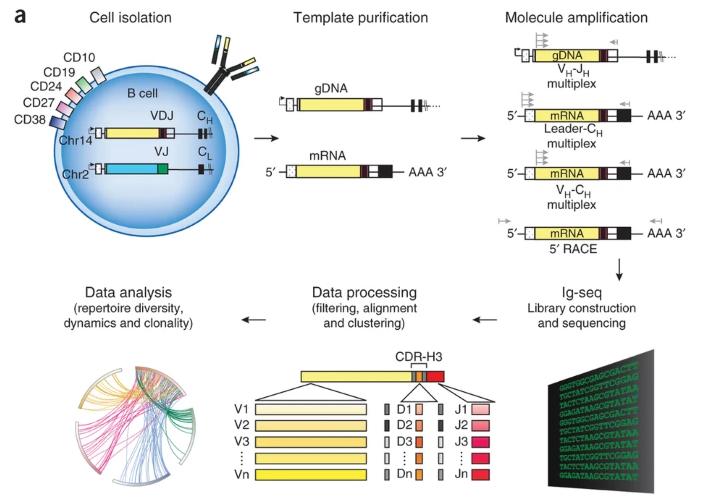 Schematic of steps involved in high-throughput sequencing of Ig genes
Schematic of steps involved in high-throughput sequencing of Ig genesWhat are Requirements for Monoclonal Antibody Drug Sequence Analysis
The confirmation of gene sequence accuracy at the levels of DNA, mRNA transcription, and protein expression constitutes the cornerstone of ensuring drug quality and patient safety. Prior to transfection of host cells, exhaustive sequencing and comparative analysis must be conducted to verify that the nucleic acid sequence of the recombinant vector fully aligns with the theoretical design. Considering the potential risk of genetic mutations during cell passage, such mutations not only exacerbate the microscopic heterogeneity of cell populations but may also confer selective advantages to mutant clones, resulting in clonal replacement. Therefore, stability studies of cell passages are of particular importance, aiming to validate the suitability of cell lines for recombinant protein drug production and ensure consistent product quality. Numerous regulatory agencies have issued explicit guidelines emphasizing the need to ensure the genetic stability of the protein expression system throughout the biopharmaceutical manufacturing process.
One of the core tasks in the evaluation of genetic stability is a comprehensive analysis of the size and nucleic acid sequence of the target region of the genome, including the identification of promoters and other key functional elements. Such analyses should cover at least the master cell bank (MCB) and the production cell bank (PCB). When submitting pharmaceutical documentation for clinical trials or marketing applications, drug development institutions are required to provide detailed sequence information at the levels of DNA, mRNA transcription, and protein expression, along with key supporting data such as gene copy number and integration sites.
From the early stages of cell line development to the full-scale commercial production process, any upstream culture process modifications must be carefully evaluated. Specifically, qualitative and quantitative studies should be conducted to address the risks associated with sequence variants resulting from genetic mutations and amino acid misincorporation, which could adversely affect the stability, biological function, and immunogenicity of the final product.
Technical Approach to Monoclonal Antibody Drug Sequence Analysis
Generally, advanced sequencing technologies are employed to assess the accuracy of genetic information and expression products from three dimensions: DNA, RNA, and amino acid sequences. The stability of these sequences is monitored throughout continuous production processes. At the molecular level, mutations in mRNA samples may arise from the DNA template, transcriptional errors, abnormal splicing, or post-transcriptional modifications (PTMs). Furthermore, improper optimization of codon usage in the exogenous gene, incorrect tRNA charging, or depletion of specific amino acids in the culture medium may result in erroneous amino acid incorporation during protein synthesis, despite the absence of genetic anomalies. Consequently, high-level technical methods, such as protein sequencing or mass spectrometry, must be employed to analyze sequences at the amino acid level, enabling a comprehensive assessment of the potential impact of sequence variants on product quality.
DNA-Level Sequence Analysis
At the DNA level, genetic stability evaluation focuses on the gene copy number, integration sites, and sequence integrity. Quantitative polymerase chain reaction (qPCR) is commonly employed to detect the copy number of exogenous genes and evaluate genetic stability. However, attention must be paid to the technical limitations of qPCR, such as differences in amplification efficiency between the reference and test samples. In contrast, digital PCR (dPCR) enhances accuracy by amplifying individual template molecules in isolation, directly counting positive and negative signals, and using Poisson distribution to calculate the absolute copy number of the target gene. This approach enables accurate absolute quantification without reliance on standard curves or reference samples.
Additionally, fluorescence in situ hybridization (FISH) serves as a powerful tool for confirming the integration sites of the target gene. Notably, during the early stages of cell line development, the timely identification and exclusion of variant clones through amplicon sequencing have successfully accelerated the development of several clinical projects.
RNA-Level Sequence Analysis
For certain multi-copy genes, transcription into mRNA may occur with varying efficiency, and sequence variations may arise during transcription. Consequently, sequence analysis of monoclonal antibody (mAb) drugs at the RNA level is particularly critical. Commonly employed analytical techniques include Northern blotting, which detects specific RNA sequences, and sequencing of cDNA following reverse transcription of mRNA, which facilitates the investigation of potential sequence changes. Moreover, RNA-sequencing (RNA-seq), an application of next-generation sequencing (NGS) at the mRNA level, enables in-depth analysis of low-frequency gene mutations.
Amino Acid-Level Sequence Analysis
Even if the mRNA sequence is confirmed to be accurate, factors such as the depletion of specific amino acids in the culture medium may lead to erroneous amino acid incorporation, resulting in the formation of sequence variants. Thus, primary amino acid sequence analysis is a critical component in structural confirmation. Typically, liquid chromatography (LC) coupled with mass spectrometry (MS) or tandem mass spectrometry (MS/MS) is employed for sample separation and analysis, commonly referred to as LC-MS or LC-MS/MS techniques. By applying various sample preparation methods, comprehensive structural information can be obtained, including molecular weight of intact antibodies, deglycosylated molecular weight, reduced molecular weight, reduced deglycosylated molecular weight, peptide molecular weight, N/C-terminal heterogeneity, glycosylation, glycation, deamidation, methionine oxidation, disulfide/sulfhydryl bonds, free thiol groups, amino acid sequence, and sequence variants.
Another noteworthy ionization technique is matrix-assisted laser desorption/ionization (MALDI), which effectively ionizes molecules with molecular weights exceeding 500 kDa, generating singly charged ions. This method is frequently employed in the study of intact molecular weight, PTMs, and modification sites. However, sequence variants generally exist at low abundance and exhibit minimal physicochemical differences from normal antibody molecules. Therefore, high-resolution, high-mass-accuracy LC-MS/MS techniques are required for both qualitative and quantitative analysis. Moreover, these methods must be appropriately validated to ensure accuracy and reliability of the results.
Select Service
Comprehensive Characterization and Analysis of Protein Structure
Building upon amino acid sequence analysis, this study focuses on the comprehensive characterization of antibody protein structure, encompassing several PTMs such as glycosylation, glycation, sulfation, and phosphorylation, along with the identification of variants including size heterogeneity, charge variants, aggregates/degradation products, and amino acid modifications like oxidation, deamidation, and cyclization. Although certain PTMs, such as glycation, N-linked oligosaccharide glycosylation, deamidation, and oxidation, can be analyzed using similar technical approaches and are closely related to the structure and activity of the antibody molecule, their effects are often influenced by upstream production processes and storage conditions.
For molecular size variants, including aggregates, fragments, and non-glycosylated heavy chains, qualitative and quantitative analyses are typically performed using size-exclusion chromatography (SEC-HPLC), sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), or capillary electrophoresis-SDS (CE-SDS). Charge variants are effectively separated and analyzed via ion-exchange chromatography (IEX-HPLC), capillary isoelectric focusing (cIEF), or capillary zone electrophoresis (CZE). Differences in hydrophobicity among antibodies and their degradation fragments are analyzed using hydrophilic interaction liquid chromatography (HILIC), hydrophobic interaction chromatography (HIC-HPLC), or reversed-phase high-performance liquid chromatography (RP-HPLC). To detect subtle differences between sequence variants and the normal antibody, often involving only one or a few amino acids, high-resolution multi-attribute methods (MAMs) are required, typically involving sample pretreatment or enrichment techniques.
A top-down approach is used to characterize the complete antibody molecule, isolating and preparing key components such as glycosylated, oxidized, charge variants, and molecular size variants to elucidate the relationship between structure and biological activity. Additionally, forced degradation studies allow for efficient preparation and identification of product-related substances and/or impurities, thereby providing crucial insights into the correlation between these elements and their structure or function. These findings contribute to the determination of critical quality attributes (CQAs) of the drug molecule and establish a robust foundation for the development of appropriate process and analytical control strategies.
The strategy of direct fragment analysis reveals critical information regarding the primary structure, glycosylation patterns, and isomeric variants of antibodies. Although the bottom-up approach requires more time, it offers more detailed structural information. In this process, protein samples are first denatured, reduced, and alkylated, followed by enzymatic digestion using proteases such as trypsin, Lys-C, or Asp-N. Subsequently, the primary structure is characterized by LC-MS/MS. Combining reversed-phase HPLC (RP-HPLC) peptide mapping with MS and MS/MS allows for accurate verification of the antibody drug's amino acid sequence as predicted from the cDNA. In the routine workflow, the molecular masses of all unmodified peptides are initially analyzed and compared with the masses predicted by MS. Peptide sequences are then further confirmed through MS/MS fragmentation. Peptide mapping, combined with MS and MS/MS, is not only utilized to determine peptide content but also to quantify the levels of modified amino acids through the integration of peak areas from the extracted ion chromatograms (EIC). The results are typically expressed as a percentage of the total peak area of the peptide.
To ensure sequence integrity and accuracy, multiple enzymatic digests are required to achieve comprehensive sequence coverage and to identify all potential amino acid modifications. It is important to note that glycation of lysine residues interferes with Lys-C digestion, necessitating the use of alternative proteases to generate complementary data. Throughout the research and development process, strict adherence to the principle of confirming amino acid sequences at both the MS and MS/MS levels is maintained. Detailed analyses of sequence coverage are conducted, along with the provision of b- and y-ion fragment information. Finally, the amino acid sequence is matched against the sequence predicted from cDNA, and in accordance with regulatory guidelines, 100% sequence coverage in primary mass spectrometry is ensured. Simultaneously, in-depth analysis of MS/MS signals is performed to uncover additional valuable insights.
Conclusion
This detailed characterization of antibody structure, encompassing molecular size and charge variants, post-translational modifications, and amino acid sequence fidelity, provides a thorough understanding of the relationship between the antibody's structure and its biological function. The multi-level analytical strategies described—top-down, bottom-up, and direct fragment analysis—enable precise verification of the antibody sequence and help to define the critical quality attributes. Such analyses are integral to the development of robust production processes and control strategies, ensuring both the efficacy and safety of monoclonal antibody therapeutics.
Reference
- Georgiou, G., Ippolito, G., Beausang, J. et al. The promise and challenge of high-throughput sequencing of the antibody repertoire. Nat Biotechnol 32, 158–168 (2014).










