Composition and Production Requirements
Antibody-drug conjugates (ADCs) consist of three primary components: an antibody, a linker, and a cytotoxic agent. The production of ADCs necessitates the separate synthesis of these components, which are subsequently conjugated. This process involves multiple complex stages and demands high precision in molecular design, preparation, and manufacturing protocols. Typically, the entire ADC production workflow includes the preparation of monoclonal antibodies (encompassing cell culture expansion, protein expression, and purification), the synthesis of linkers and small molecule drugs, conjugation, and final product formulation. Due to the distinctive attributes of antibody drugs, attention must be directed not only to the quality attributes, process characteristics, and process control of the antibodies but also to the manufacturing environment, including facility, equipment, and contamination control.
Fundamental Requirements and Principles of ADC Production
The ADC production process typically comprises several stages: monoclonal antibody preparation (including cell expansion, protein expression, and protein purification), linker and small molecule drug synthesis, conjugation, and final product formulation. Given the unique properties of antibody drugs, it is imperative to focus on the quality attributes of the antibody, process characteristics, and process control. Additionally, significant attention must be given to the manufacturing environment, encompassing facilities, equipment, and contamination control.
To ensure the reliability and reproducibility of the ADC manufacturing process, a thorough risk assessment is required prior to market release. Experimental design (Design of Experiments, DoE) should be employed to identify and control critical process parameters that influence drug quality attributes. By determining an appropriate design space, the manufacturing process can be optimized to ensure smooth operation and maintain the controllability of ADC product quality.
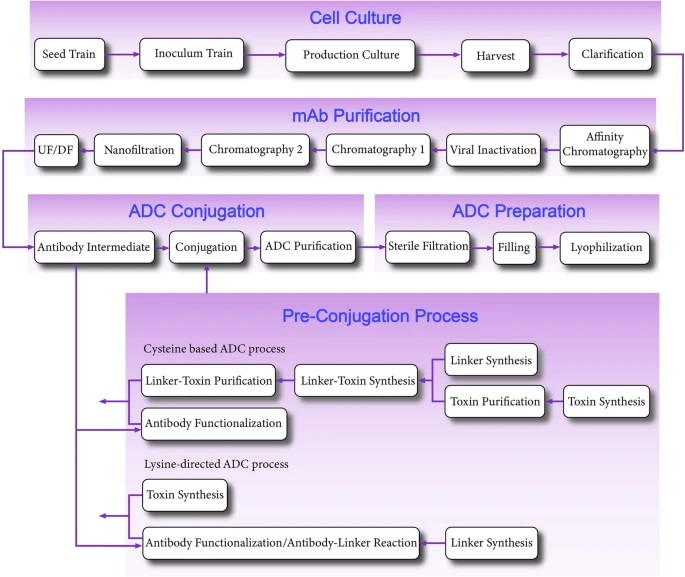 ADC production process flow chart
ADC production process flow chartThe production of ADCs involves a multifaceted biotechnological and chemical synthesis process, adhering to stringent procedural and quality standards. The following outlines the critical stages of ADC production:
Monoclonal Antibody (mAb) Production The production of monoclonal antibodies (mAbs) follows a conventional biomanufacturing protocol, encompassing both cell culture and purification stages. Initially, mammalian cell lines, such as Chinese hamster ovary (CHO) cells, are cultured in bioreactors to express the desired antibodies. Subsequent purification employs a series of chromatographic techniques, including protein A affinity chromatography, ion exchange chromatography, and size exclusion chromatography, to ensure high-purity mAb suitable for further processing.
Preparation of Antibody Intermediate (AI) The antibody intermediate (AI) is prepared by conjugating the purified mAb with drug-linker compounds. These compounds can be synthesized chemically or produced via biotechnological methods. The conjugation process typically involves a series of well-defined chemical reactions.
Conjugation Process The conjugation process consists of several critical steps:
Synthesis of Linker-Drug Complex: The linker-drug complex, often comprising a cysteine-based linker, is synthesized.
Reaction with mAb: The mAb is subjected to a chemical reaction with the linker-drug complex. This can be a direct reaction or a sequential process where the mAb first reacts with the linker, followed by attachment to the drug molecule. For instance, lysine-directed conjugation is commonly utilized to achieve site-specific attachment.
Purification of ADC Drug Substance Following the conjugation, the resulting ADC drug substance undergoes rigorous purification. This purification involves multiple chromatographic steps and filtration techniques to remove unconjugated drug molecules, free linkers, and other impurities. The goal is to obtain a homogenous ADC preparation with defined DARs.
Final Drug Product Manufacturing The purified ADC drug substance is then formulated into the final drug product. This step includes:
Aseptic Filling: The ADC is filled into sterile containers under aseptic conditions to maintain product sterility.
Lyophilization (if applicable): For enhanced stability and shelf-life, the ADC may undergo lyophilization, a freeze-drying process that removes water by sublimation under low temperature and vacuum conditions.
Quality Control and Packaging The final ADC product undergoes comprehensive quality control testing to ensure compliance with pharmacopeial standards and regulatory requirements. This includes assays for potency, purity, sterility, and stability. Once verified, the product is packaged and prepared for distribution.
Production of Monoclonal Antibodies
The production of monoclonal antibodies entails requirements similar to other recombinant monoclonal antibody products in the process of ADC manufacturing. Critical process parameters and ranges must be determined based on the product's critical quality attributes (CQA) to ensure consistency and traceability of the production process.
During the production process, it is essential to establish relevant quality control specifications covering raw materials, excipients, and critical materials to ensure consistency and controllability between different batches.
Compliance with relevant regulations is necessary for the sourcing, management, and testing of engineered cell lines. Additionally, stringent monitoring of impurities and removal of infectious agents related to the process is crucial. It is important to avoid degradation, aggregation, or instability of monoclonal antibodies during non-site-specific conjugation, keeping stability controlled at very low levels. Mismatches in site-specific conjugation techniques may result in structural alterations of the monoclonal antibody protein. Therefore, these issues need to be thoroughly considered early in the design of monoclonal antibodies for ADCs.
Utilizing control strategies established during product process development and process characterization, specific monoclonal antibodies are produced using recombinant engineered cell lines for recovery, amplification, and production cultivation expression. Key parameters control during cell culture is crucial. Initial purification steps for monoclonal antibodies typically involve affinity chromatography. Low pH treatment is a common method for virus inactivation. If the product is unstable under low pH conditions, alternative virus inactivation methods may be employed. Moderate and fine purification typically involves ion exchange chromatography, hydrophobic interaction chromatography, and mixed-bed chromatography to remove process and product-related impurities from the protein. Most purification processes also include nanofiltration steps to remove small viruses resistant to chemical inactivation. Finally, ultrafiltration is used to concentrate monoclonal antibodies and alter the buffer.
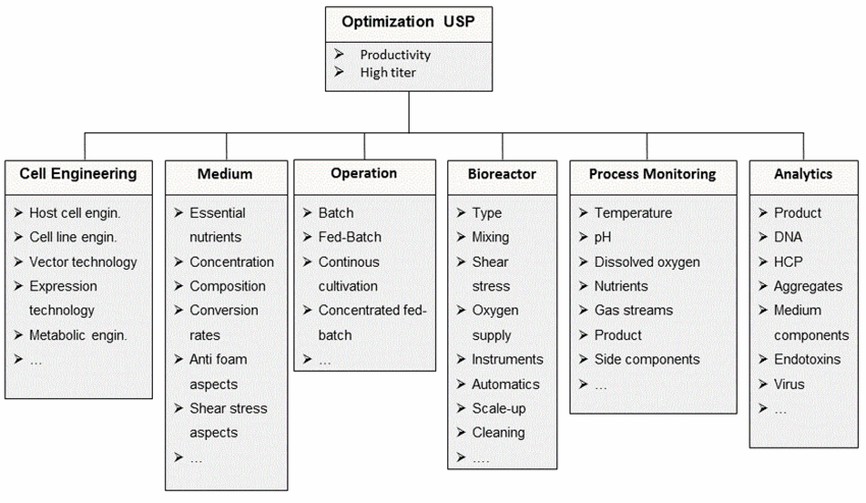 Optimization process of upstream production process of monoclonal antibody products
Optimization process of upstream production process of monoclonal antibody productsProduction of Conjugates and Small Molecule Drugs
In order to determine the impact on critical quality attributes, key process steps and process parameter ranges for the preparation of conjugates should be identified. Based on an understanding of the product and process, quality control strategies for intermediates, bulk products, and final products should be gradually established to ensure consistency between batches.
The production of small molecule drugs should be based on scientific knowledge and principles of risk management, developing control strategies for binding impurities, chiral impurities, genotoxic impurities, and other impurities. The limits for impurity control should be established based on the research results of impurity removal in subsequent processes.
Quality control specifications for intermediates, key raw materials, and reagents should be established. For chiral materials, the limits for enantiomers or diastereomers should be controlled. Process parameters affecting product quality during preparation should be controlled (such as feed rates, process temperatures, drying temperatures). When impurities in raw materials or intermediates are related to critical quality attributes of the product, the ability of the production process to remove these impurities or their derivatives should be validated. For processes involving the recycling of fillers, attention should be paid to filler lifespan and cleaning methods, supported by relevant research.
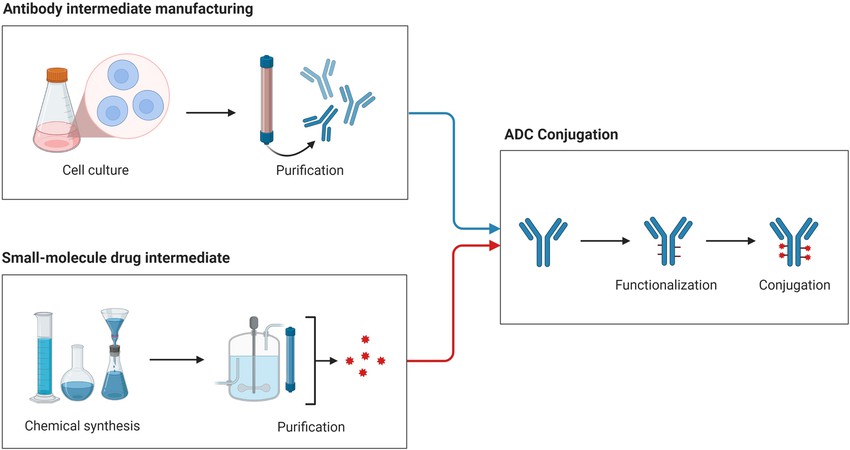 ADC development, purification and production
ADC development, purification and productionADC Conjugation Process Requirements
Development and Control Strategies of ADC Conjugation Processes
The conjugation methods of ADCs are primarily categorized into non-site-specific and site-specific conjugation. Non-site-specific conjugation presents significant challenges due to heterogeneity arising from variations in drug load and conjugation sites. Therefore, it is imperative to meticulously control the antibody functionalization steps to ensure batch-to-batch consistency. Conversely, site-specific conjugation employs genetic engineering techniques to introduce specific binding sites within the antibody structure, facilitating the production of homogenous ADC products.
Similar to the process requirements for monoclonal antibodies, the conjugation process must consider various parameters of ADCs. Given that antibodies are hydrophilic and cytotoxic drugs are typically hydrophobic, organic solvents are employed to dissolve both entities for covalent conjugation. However, organic solvents may induce antibody aggregation, making the selection of appropriate solvents crucial for successful conjugation. Additionally, the determination of process parameter ranges, including antibody concentration, initial material quality, antibody functionalization conditions, linker-to-drug ratio, and conjugation time, is essential for the reproducibility of the ADC conjugation process.
The conjugation process is critical to the success of ADCs. Reaction parameters must be controlled according to the specific type of reaction, with appropriate process controls established. Optimal process conditions and control ranges should be determined through accumulated data. Furthermore, it is essential to control the production environment of cytotoxic drugs to prevent health risks to personnel and contamination of equipment, thereby maintaining the biological load and endotoxin levels of the product.
Selection and Verification of Critical Starting Materials
The critical starting materials utilized during the ADC conjugation process include monoclonal antibodies and small molecules (linkers and toxins) obtained from upstream ADC production. These materials are foundational to ADC production, significantly influencing product quality and process stability. Therefore, it is imperative to establish internal control standards for these starting materials.
A comprehensive investigation and control of potential impurities introduced by starting materials and production reagents are necessary. Relevant impurity standards should be procured or prepared for impurity profiling. For starting materials with chiral structures, precise monitoring of enantiomers and diastereomers is required. It is essential to identify impurities potentially introduced during the production process of these starting materials. During process development, the selection of low-toxicity reagents and solvents should be prioritized, and appropriate residual control limits must be established.
Selection and Verification of Production and Process Development Equipment
From a safety and microbiological control standpoint, it is recommended that the active pharmaceutical ingredient (API) for ADCs be produced in a negative-pressure isolator. Strict adherence to maintaining the airtightness of the isolator is crucial. In large-scale production settings, the isolator can be linked to reaction vessels which are configured as advanced biosafety laboratory isolation systems. It is imperative that the high-efficiency particulate air (HEPA) filters utilized in the production equipment be readily replaceable to prevent the release of any potentially harmful substances into the ambient surroundings.
The reactor vessel is a critical chemical reaction container in ADC production. Reaction temperature significantly influences the quality of the ADC product. Therefore, reactor vessels capable of precise temperature control should be selected. Ultrafiltration systems are also essential in the production of biological products, with tangential flow filtration being preferred over traditional dead-end filtration due to its continuous and stable characteristics.
Equipment and facilities should be capable of clean-in-place/sterilize-in-place (CIP/SIP) operations. The use of reaction equipment should be product-specific to avoid cross-contamination. If equipment sharing is unavoidable, cleaning validation should focus on the presence of coupling residues and the associated risks.
You may interested in
Learn more
Development and Control Strategies for Purification Processes
Following the conjugation of linkers and small molecule drugs, it is imperative to purify the resultant ADC to remove product-related and process-related impurities. Typically, conjugated ADCs undergo purification via ultrafiltration, frequently utilizing continuous flow systems such as tangential flow filtration (TFF). The purification process steps must be appropriately designed based on the characteristics of the reagents, redox agents, and chemical conjugation agents used. The process must effectively eliminate residual unconjugated antibodies, small molecule drugs, linkers, and linker-drug conjugates. Efficient removal of high-molecular-weight and low-molecular-weight impurities is essential to ensure product stability within acceptable limits.
Preventing microbial contamination during the purification process is crucial. Depending on the conjugation method, it may be necessary to purify certain intermediates or exchange buffer solutions. The selection of operational parameters is critical for effective impurity removal. Parameters such as flow rate, transmembrane pressure, membrane manufacturer and quality, purification yield, purification time, and other relevant factors must be thoroughly investigated and validated. Additionally, the impact of exposure time and temperature fluctuations during the ADC purification process on product stability should be rigorously studied.
Development and Control Strategies for Pharmaceutical Processes
Current ADC products, whether under development or already marketed, typically adopt lyophilized or liquid formulations. The formulation of ADCs should be based on a comprehensive understanding of the quality attributes of both the ADC product and the monoclonal antibody. Additionally, the stability of the small molecule drug and the linker must be considered. The intended route of administration and clinical compatibility should be determined as early as possible.
The primary components of ADC formulations generally include pH buffers, sugars, and surfactants. The function of each component in the formulation is determined by the physicochemical instability of the ADC protein and the degradation mechanisms of the linker and small molecule drug. Key considerations include pH, protein concentration, stabilizers, surfactants, and the selection of packaging containers. These components must meet the requirements for product storage, transportation, and clinical use.
The development of pharmaceutical processes involves the study and optimization of process parameters to identify critical process parameters. Process robustness studies establish the operational ranges, followed by the definition of control specifications and acceptance criteria. Finally, process validation is conducted to assess the reproducibility of product quality. The primary control points in pharmaceutical processes typically encompass environmental control during production, control of critical process parameters, and quality control of intermediates. Appropriate process conditions and parameters ensure the reproducibility of the manufacturing process, facilitate the quality control of intermediates, and guarantee the quality of the final product.
Risk-Based Extractables and Leachables Studies
The use of numerous single-use components throughout the ADC manufacturing process necessitates the simplification of extractables and leachables (E&L) studies to reduce costs and expedite time-to-market. This approach should be grounded in scientific principles and risk assessment methodologies. Relevant guidelines, such as USP <665> and USP <1665> draft guidelines, the BioPhorum Operations Group (BPOG) documents, and ICH Q3 and M7, can serve as references for establishing appropriate risk assessment models. Key risk dimensions to consider include contact duration, contact temperature, chemical composition of process fluids, chemical composition of process components, clearance capabilities of subsequent processes, and clinical usage information (e.g., dosage form, administration cycle, and daily dosage). Ultimately, E&L study strategies and corresponding toxicity and safety evaluations should be implemented based on the risk assessment outcomes.
Extractables and leachables can adversely impact the final product's quality, potentially leading to secondary safety and immunogenicity issues. For instance, tungsten can induce protein denaturation and aggregation, thereby triggering immunogenicity. Evaluating and understanding potential immunogenicity issues remain challenging because the correlation between the properties and concentrations of leachables and their effects has not been well established. Consequently, predicting safety and immunogenicity risks based solely on extractables and leachables data is difficult. Therefore, traditional compatibility experiments, such as stability and safety studies, are necessary for comprehensive assessment.
Particular attention must be given to solvents commonly used in the ADC conjugation process, such as dimethyl sulfoxide (DMSO) and N,N-dimethylacetamide (DMAC). These solvents exhibit high extractive capacities at elevated concentrations and pose compatibility risks with single-use components like polyethersulfone (PES) and polyvinylidene fluoride (PVDF) filters and sensors. Therefore, extractables studies tailored to specific solvent models should be integrated with the actual process components used in these process steps. Additionally, during the conjugation process, both the exposed antibodies and the linker-toxin complex exhibit high reactivity, necessitating attention to potential reaction byproducts formed between leachables and the linker, toxin, and antibody.
Quality Control of Antibody-Drug Conjugates
Quality control strategies encompass a series of planned controls derived from the current understanding of the product and process to ensure process performance and product quality. These specifications are part of an overall control strategy, which includes a list of tests, analytical procedure references, and appropriate acceptance criteria. The selection of fundamental specifications aims to confirm the comprehensive quality of the active pharmaceutical ingredient (API) and the drug product, rather than to establish a complete characterization. Therefore, developing appropriate specifications requires a systematic approach.
The methodology for establishing commercial specifications for biotherapeutic products and impurities has been well-established and widely recognized within the industry. This approach emphasizes a risk-based strategy grounded in product knowledge to ensure patient safety and efficacy. A systematic method has been published for identifying and evaluating CQAs. These CQAs include product variants, process-related impurities, obligatory CQAs, raw materials, and leachable compounds. Characterizing the product at the levels of the antibody, small molecule, or raw materials using representative materials is essential to gain a comprehensive understanding of the physicochemical and biological properties of the product. Chromatographic and electrophoretic techniques, spectroscopic methods, and mass spectrometry are extensively employed for physicochemical characterization, while binding assays, ELISA (enzyme-linked immunosorbent assay), and cell-based bioassays are widely utilized for biological characterization.
By integrating these methodologies, a robust quality control framework can be established, ensuring that the ADC meets the required standards for safety, efficacy, and consistency, thereby safeguarding patient health and complying with regulatory requirements.
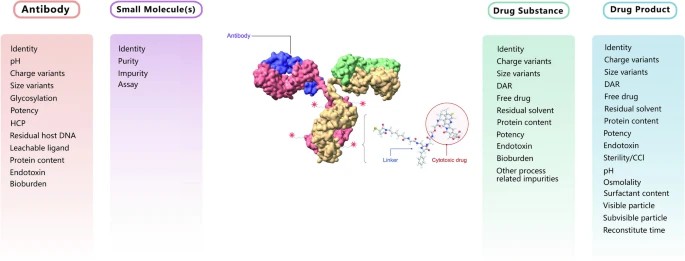 Typical CQAs for Antibodies, Small Molecules, APIs, and Drug Products
Typical CQAs for Antibodies, Small Molecules, APIs, and Drug ProductsMaintaining batch-to-batch consistency after scale-up presents a significant challenge in process development. Key parameters include the average DAR, conjugate distribution ratio, purity, and residual impurities. Achieving a stable and controllable process necessitates extensive scientific risk assessments and experimental work. The stability of reaction reagents and intermediates also critically impacts process performance and product quality. Therefore, it is essential to conduct thorough studies and establish appropriate control strategies, such as selecting suitable reducing agents and solvents and controlling storage durations. Post-conjugation steps such as diafiltration or quenching should be promptly implemented.
DAR
The DAR directly influences the safety, efficacy, and pharmacokinetics (PK) of the product. During the production of the drug substance (DS), the DAR is controlled and tested using appropriate analytical methods. Further evaluation of DS stability and the impact of the drug product (DP) process on DAR is required to determine the necessity of DAR testing at these stages. Acceptance criteria are largely determined by clinical experience specific to the product.
Current production processes and quality control methods commonly employed include ultraviolet-visible spectroscopy (UV-Vis), hydrophobic interaction chromatography (HIC), reverse-phase liquid chromatography (RPLC), and size-exclusion chromatography-mass spectrometry (SEC-MS) for DAR characterization.
UV-Vis Method: This method measures the DAR by exploiting the differential maximum absorption wavelengths of the drug-linker and the monoclonal antibody. However, it does not provide information on DAR distribution and is susceptible to interference from free drugs.
HIC: This technique separates different DAR molecules based on hydrophobicity differences and provides analytical information on DAR and DAR distribution. It is commonly used for native analysis of cysteine-conjugated ADCs. Nonetheless, it has limited resolution for samples with high DAR.
RPLC: Under denaturing conditions, RPLC separates different DAR molecules or related subunits, generally offering better resolution than HIC. However, cysteine-conjugated ADCs dissociate into light chains and heavy chains under RPLC conditions, yielding only the average DAR and drug distribution on these chains.
SEC-MS: This technique exhibits excellent resolution for different DARs and is compatible with most ADCs. Consequently, SEC-MS is increasingly utilized in early development and characterization of ADCs.
You may interested in
Learn more
Distribution of Conjugation Sites and Unconjugated mAb Portions
ADC products exhibit heterogeneity, comprising molecules with varying numbers and sites of conjugation, which may impact their safety and efficacy. The degree of heterogeneity depends on the ADC platform, with lysine-directed, interchain cysteine-directed, and site-specific conjugation platforms ranked in descending order according to current knowledge. The distribution of conjugation sites and unconjugated mAb portions can be correlated with the DAR for product characterization and controlled characterization through DAR testing.
Free Drug
Free drugs represent unconjugated drugs with potential systemic toxicity. While free drugs may exist in various forms, such as drug or linker-drug, they are typically tightly controlled. Acceptance criteria for free drugs are established based on their toxicity, taking into account process clearance capabilities. Control of free drugs is necessary during DS release, and testing at these stages may also be required based on the stability of ADC products during DS storage, DP processes, and DP storage.
Residual Solvents
Organic solvents are commonly used in DS production to dissolve hydrophobic cytotoxins. Acceptance criteria for residual solvents should be established based on solvent toxicity and process removal capabilities. Residual solvent testing typically occurs during DS release, with testing requirements at other stages determined based on the impact of residual solvents on DS stability, DP processes, and DP stability.
Size Variants
Size variants typically include fragments and aggregates, often existing in dissociable and non-dissociable forms. Acceptance criteria for DP at the end of shelf life should ensure efficacy and patient safety, primarily based on product-specific clinical experience. Suitable acceptance criteria should be established for antibodies, DS, and DP stages based on process and stability impacts. Low molecular weight forms (LMW) can form through peptide bond or interchain cleavage. Similar strategies can be employed to establish appropriate LMW standards for aggregates, especially considering ADC fragments may carry conjugated drugs, potentially leading to off-target toxicity. Therefore, LMW forms should be characterized and controlled below safe levels.
Charge Variants
Charge variants are common in antibodies and ADCs. Modifications (including deamidation, aspartate isomerization, glycation, and C-terminal lysine) can lead to charge transfer and charge isoform variation. Each modification should be individually assessed based on clinical experience for cost and appropriate acceptance criteria set.
You may interested in
Learn more
Other Forms of Variants
Several variants need consideration depending on product characteristics and ADC platforms. These include but are not limited to oxidation-induced variants, sequence variants, free disulfide bonds, and thioether bonds. These attributes may affect product safety or efficacy and can impact ADC product quality. For example, disulfide bonds may not be a CQA for mAbs but are crucial for interchain cysteine ADC platforms due to their effect on DAR. These variants should be evaluated on a case-by-case basis.
Quantity or Potency
Protein content is typically included in DS and DP formulations to ensure accurate dosing of the product.
Potency
Potency assays should be defined through a reflective action. For ADC products, they are primarily developed to kill tumor cells within cancer patients. Cell-based bioassays are anticipated for potency testing. These bioassays should demonstrate stability, as proven by method validation. Variability in methods should be considered to set appropriate acceptance criteria.
Glycosylation Modifications
Glycosylation patterns may affect efficacy, safety, or PK. For ADC products, the primary function of the mAb is to deliver the drug to target cells, thus glycosylation may not affect efficacy. ADC products should be thoroughly characterized to prevent secondary mechanisms of action, which may be influenced by glycosylation patterns. In such cases, glycosylation can be evaluated as a CQA and included in AI IPC or release testing based on process capability.
You may interested in
Linker, Drug, or Linker-Drug
Quality control of small molecule drug intermediates is similar to that of active pharmaceutical ingredients of small molecule drugs, particularly considering their impact on ADC product.
Purity
Purity is a typical CQA. Broader acceptance criteria depend on its impact on ADC product quality and DS processes. If the addition of excess linkers or linker drugs does not affect DAR and can be effectively removed through the process, acceptance criteria are reasonable.
You may interested in
Conjugatable Impurities
Conjugatable impurities possess reactive functional groups, enabling them to react with AI to generate by-products. These by-products are challenging to remove as these proteinaceous materials are difficult to separate from ADC products. Therefore, conjugatable impurities are controlled and tested during small molecule manufacturing stages.
Non-Conjugatable Impurities
Non-conjugatable impurities lack functional groups to conjugate with monoclonal antibodies. These impurities are controlled at lower levels to minimize potential risks to patients. Control and testing of these impurities occur during small molecule stages.
Other Process-Related Impurities
Other process-related impurities may enter the final product from various sources, such as residual heavy metals in DS processes. Each impurity should be individually evaluated to determine CQA. These impurities should be controlled below safe levels based on clinical experience, prior knowledge, or regulatory requirements.
Process capability is a key consideration in setting testing strategies. If the process has been demonstrated to effectively remove impurities, quality control testing will add minimal value.
Surfactant Content
Surfactants are typically added to formulations to protect products and enhance stability. Acceptance criteria for surfactant content are established based on formulation development data. Testing strategies depend on the manufacturing process and stability of surfactants under storage conditions.
Other Quality Attributes
There are additional quality attributes, such as pharmacopoeial requirements, including color, clarity, endotoxin levels, sterility, visible particles, sub-visible particles, pH, osmolarity, extractable volume, fill weight of solid dose, reconstitution time, and moisture. Acceptance can be set based on pharmacopoeial requirements or formulation development. Modern process controls may be more robust than final product testing. An example is online fill weight, which provides stronger process control than final product fill weight or extractable volume testing.
Extractables and Leachables
Extractables may originate from different sources, such as manufacturing processes or container closure systems, and include organic and inorganic impurities as elemental impurities. These are typically characterized under process representative conditions or storage conditions to demonstrate impurity control at safe levels. ICH guidelines, such as the ICH Q3 series, provide detailed information on safety levels. Extractables may or may not be included in AI, DS, or DP testing as their levels are typically well-controlled.
References
- Li, Meng, et al. "Antibody-Drug Conjugate Overview: a State-of-the-art Manufacturing Process and Control Strategy." Pharmaceutical Research (2024): 1-22.
- Marei, Hany E., Carlo Cenciarelli, and Anwarul Hasan. "Potential of antibody–drug conjugates (ADCs) for cancer therapy." Cancer Cell International 22.1 (2022): 1-12.











