Primary Objectives of Phase I Clinical Trials
Phase I clinical trials constitute a pivotal juncture in the development of novel therapeutic agents. The primary aim of these trials is to evaluate the safety and pharmacokinetic (PK) properties of a new drug in humans, following successful preclinical pharmacological and toxicological assessments in animal models. This phase marks the initial administration of the drug in human subjects with the foremost goal of assessing the drug's tolerability and PK profile in a clinical environment.
In this initial phase, dose escalation is systematically performed, while vigilant monitoring and documentation of any adverse effects are conducted. The PK parameters—including absorption, distribution, metabolism, and excretion—are thoroughly investigated. Concurrently, preliminary data regarding the pharmacodynamic (PD) effects—such as the therapeutic or biological response—may also be gathered.
Specifically, Phase I trials aim to determine the maximum tolerable dose (MTD) and the dose-limiting toxicity (DLT) of the investigational drug. Tolerability is assessed through careful observation of PK properties, while PD effects are analyzed to understand the drug's impact on biological systems.
The data and insights gained from Phase I trials are vital for the design of subsequent Phase II and Phase III clinical trials, as they inform dosing regimens and safety protocols. Ultimately, Phase I trials lay a robust foundation for ensuring the safety and efficacy of the drug in future clinical studies.
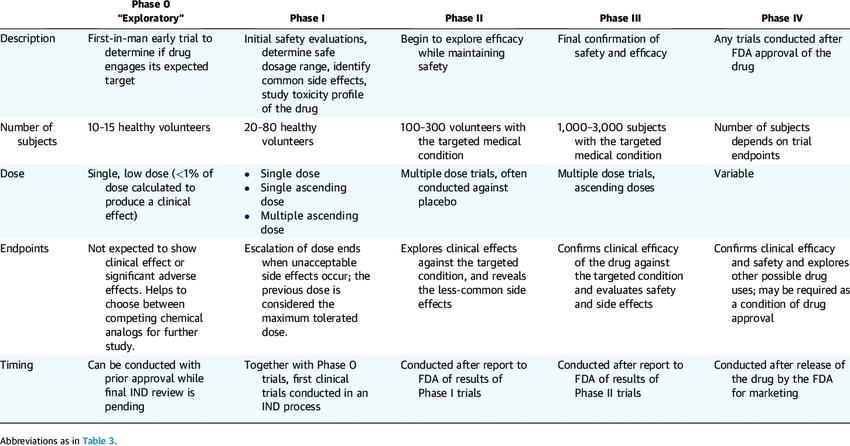 Characteristics of Clinical Trial Phases (Gail Van Norman 2016)
Characteristics of Clinical Trial Phases (Gail Van Norman 2016)Primary Focus of Phase I Clinical Trials
Core Research Questions
Phase I clinical trials primarily aim to address two key questions:
1. What adverse reactions may the drug cause?
2. How is the drug absorbed, distributed, metabolized, and excreted in humans?
To comprehensively answer these questions, a series of critical investigations must be completed. Moreover, Phase I trials often incorporate concurrent assessments of pharmacodynamics (PD) in humans, evaluating factors such as the influence of food on drug efficacy and potential drug-drug interactions to obtain a broader understanding of the drug's characteristics.
Select Service
Organized Execution of Phase I Clinical Trials
Trial Sequence
Phase I clinical trials typically follow a structured sequence of steps. These include initial single-dose tolerability studies, followed by single-dose pharmacokinetic (PK) studies, and concluding with multiple-dose tolerability and PK studies. Notably, when Phase I trials involve patient populations, tolerability and PK assessments may be conducted in parallel to improve trial efficiency.
Design Principles
From a design perspective, Phase I trials often employ an open-label, self-controlled format. However, in cases where primary adverse reactions lack clear objective markers or when the relationship between adverse effects and the drug is not readily identifiable, randomized, placebo-controlled, and blinded designs are favored to enhance rigor and reliability.
Participant Selection
Healthy, young male volunteers are typically preferred as participants in Phase I trials. However, for specific classes of drugs, such as cytotoxic agents (e.g., antineoplastic drugs), patient populations are mandated to ensure the clinical relevance and applicability of the trial outcomes.
Sample Size Determination
The sample size for Phase I trials generally ranges from 20 to 80 subjects, balancing the representativeness and feasibility of the trial while ensuring the robustness of the conclusions drawn.
Design of Human Tolerability Studies in Phase I Clinical Trials
Human tolerability studies are designed to evaluate the tolerability of different drug doses in humans, with the primary goal of identifying the nature of adverse reactions and their corresponding dose ranges. Typically, Phase I trials begin by exploring the tolerability of a single dose, followed by potential progression to multiple-dose studies based on preliminary findings.
Several strategies can be employed in the design of these studies, including open-label trials, baseline-controlled trials, and randomized, blinded trials to enhance the accuracy and reliability of observed outcomes. The primary objective is to minimize bias and ensure the scientific validity and reliability of the trial results.
The overall design concept of human tolerability studies in Phase I trials is to gradually escalate the dose from an initial starting dose to the maximum dose, with multiple intermediate dose groups established along the way. Each dose group follows a predetermined dosing protocol, systematically exploring the dose-response relationship until either the maximal tolerable dose (MTD) is identified or the pre-specified dose limit is reached. This methodical approach provides critical safety and efficacy data that will inform subsequent stages of drug development.
Determining the Maximum Dose in Phase I Tolerability Studies
Several factors should be considered when determining the maximum dose for Phase I tolerability studies:
- The maximum single dose of the same or similar drug, or drugs with a similar chemical structure, used in prior studies.
- One-tenth of the dose that caused toxic effects or reversible organ changes in long-term toxicity studies in female animals.
- One-fifth to one-half of the maximum tolerable dose observed in long-term toxicity studies in female animals.
The maximum dose design should include the expected therapeutic dose within this range. If no adverse reactions are observed upon reaching the pre-specified maximum dose, the trial may continue until the predetermined endpoint. However, if pre-specified termination criteria or serious adverse reactions occur before reaching the maximum dose, the trial should be immediately halted.
This rigorous and scientific approach ensures that the maximum dose is safely and effectively determined, providing valuable data for the drug's further clinical development.
Considerations for Protein and Antibody Drug Characterization in Phase I Trials
In the context of Phase I clinical trials involving protein-based therapeutics or monoclonal antibodies, additional considerations are imperative for drug characterization. Unlike small-molecule drugs, these biologics pose unique challenges due to their immunogenicity and complex pharmacokinetic profiles, which stem from their size, structure, and interactions with the immune system.
Characterizing protein and antibody drugs entails several critical analyses, including binding specificity, structural integrity, and in vivo stability. Furthermore, understanding the pharmacodynamic relationships between dose and biological response is crucial in the early stages to optimize dosing strategies and mitigate potential adverse effects.
Bispecific antibodies and antibody-drug conjugates (ADCs) present a need for comprehensive characterization due to their multifunctional nature and complex drug metabolism pathways. These biotherapeutics are subjected to detailed examination to ensure that their pharmacokinetics and pharmacodynamics are well-understood before advancing to Phase II trials. These assessments, integrated within Phase I clinical research, enhance the understanding of the therapeutic index and guide subsequent development phases, ensuring the drug's safety and efficacy.
Incorporating robust protein and antibody drug characterization into Phase I trials enables researchers to address the complexities of biologic therapeutics more effectively, thus supporting a more informed and efficient drug development process.










