Early Clinical Trial Research
Early-stage clinical trials, often termed pharmacological studies, primarily aim to accumulate critical data on drug safety and tolerability. When preparing for First-In-Human (FIH) trials, a well-designed protocol is of paramount importance for ensuring safety and guiding subsequent research decisions. This phase offers an initial opportunity to assess the pharmacokinetics (PK) and potential pharmacodynamic (PD) effects of the investigational compound in humans. Although most FIH trials utilize randomized, double-blind, placebo-controlled designs, many practical decisions surrounding trial protocol remain heavily dependent on preclinical data. These decisions may encompass determining whether the compound should be tested in healthy volunteers or patient populations, selecting appropriate dose groups, deciding on the number of participants, sequencing trial stages, establishing the starting dose and its escalation strategy, setting follow-up intervals, and considering whether integrated protocols for additional drug effect evaluations are warranted.
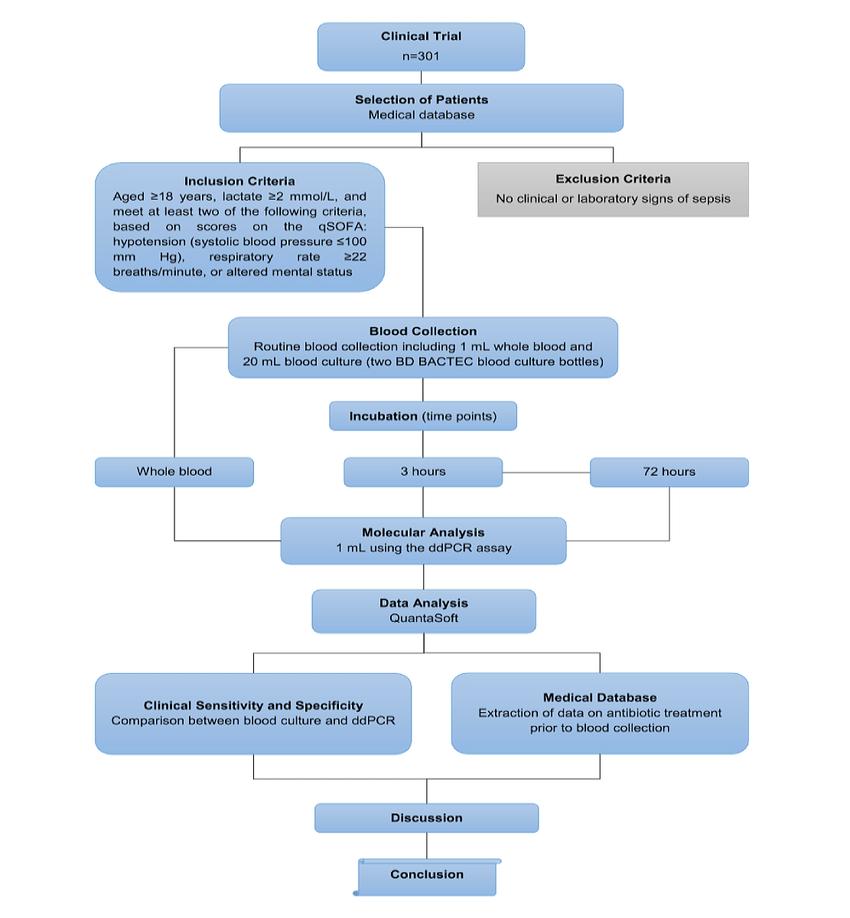 Flowchart showing the study design of the clinical trial in phase 3
Flowchart showing the study design of the clinical trial in phase 3Clinical Research Process for Anticancer Drugs
The clinical research process for anticancer drugs typically involves three phases: Phase I, Phase II, and Phase III trials. Phase I trials focus on drug tolerability and initial pharmacokinetic studies, laying the foundation for dosing regimen design in subsequent trials. Phase II trials are more exploratory, concentrating on dose optimization, preliminary efficacy evaluation, and continued safety monitoring. Phase III trials, built upon Phase II findings, aim to confirm the clinical benefits for cancer patients, providing robust evidence to support the drug's marketing approval. As the critical intermediary, Phase II trials play a pivotal role in eliminating ineffective compounds and identifying sensitive tumor types, which directly informs the decision-making for Phase III trial designs.
Key Components of Phase III Clinical Trials
The primary objective of Phase III clinical trials is to establish the benefit-risk profile of a novel therapeutic. To achieve this, Phase III trials commonly adopt randomized, double-blind, controlled designs with a sufficiently large sample size to ensure statistical power. Moreover, Phase III trials may involve dose-response studies and drug-drug interaction research based on the drug's characteristics and target patient population. Upon trial completion, statistically significant conclusions must be provided, including information on the drug's indication, target disease population, primary efficacy endpoints, dosing regimens, and safety profiles. This comprehensive dataset supports regulatory submissions and informs the risk-benefit analysis. Depending on the indication or combination therapy requirements, sponsors may further subdivide Phase III trials into Phase IIIa and IIIb trials. Following the successful conclusion of Phase IIIa, sponsors can apply for marketing approval, accelerating the time-to-market and enhancing revenue potential. Phase IIIb trials, in turn, allow for the expansion of the drug's indications, increasing market reach.
Conducting Phase III Clinical Trials
Trial Design Principles
Phase III clinical trials typically involve a comparison of the investigational drug with standard-of-care treatments and can be classified into superiority and non-inferiority trials. These trials often employ randomized, double-blind, positive-control designs. In cases where no approved positive-control drugs exist, placebo-controlled studies may be utilized.
Study Population
Participants in Phase III clinical trials are usually selected from patient populations that match the target indication of the investigational drug.
Sample Size
The sample size of Phase III clinical trials generally ranges from several hundred to several thousand participants, ensuring both the reliability and representativeness of the trial outcomes.
Overview of Phase III Trial Design
Phase III clinical trials frequently adopt randomized, parallel-control designs to determine the efficacy and safety of the investigational drug in a specific patient population. The careful selection of an appropriate trial design is critical, as it directly impacts sample size estimation, study workflow, and quality control measures. Consequently, researchers must flexibly choose the most suitable trial design based on the trial's specific objectives and existing conditions.
Select Service
Structural Characterization of Protein or Antibody Drugs in Phase III Clinical Trials
Structural characterization of protein and antibody drugs is a critical part of drug development, especially in Phase III clinical trials, where the focus is on ensuring consistency, safety, and efficacy before regulatory approval. This process involves a comprehensive set of analytical techniques to determine the molecular identity, stability, and quality of the biologic. Here's an overview of the key elements involved:
1. Primary Structure Characterization
- Amino Acid Sequence Analysis: The exact sequence of amino acids is crucial for the function of protein and antibody drugs. Mass spectrometry (MS) and Edman degradation are commonly used techniques.
- Post-Translational Modifications (PTMs): Modifications such as glycosylation, phosphorylation, oxidation, and deamidation can impact the drug's function and stability. Mass spectrometry is essential to detect and quantify PTMs.
2. Higher-Order Structure (HOS) Analysis
- Secondary Structure: Circular dichroism (CD) and Fourier-transform infrared spectroscopy (FTIR) are used to analyze α-helices and β-sheets in proteins.
- Tertiary Structure: X-ray crystallography, nuclear magnetic resonance (NMR), and small-angle X-ray scattering (SAXS) are employed to determine the 3D structure of the drug.
- Quaternary Structure: For antibodies, the arrangement of subunits (heavy and light chains) is critical. Analytical ultracentrifugation (AUC) and size-exclusion chromatography coupled with multi-angle light scattering (SEC-MALS) are used for this purpose.
3. Aggregation and Stability Studies
- Aggregation: Protein aggregation can affect drug efficacy and safety (immunogenicity). Techniques such as dynamic light scattering (DLS), SEC-MALS, and analytical ultracentrifugation (AUC) are used to detect and quantify aggregates.
- Stability Testing: Thermal stability is assessed using differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA). Forced degradation studies are also conducted to understand the behavior of the drug under stress conditions (temperature, pH, and light).
4. Glycosylation Profiling (for Antibodies and Glycoproteins)
- Glycosylation is a critical quality attribute for monoclonal antibodies (mAbs) as it affects pharmacokinetics, efficacy (e.g., antibody-dependent cellular cytotoxicity, ADCC), and immunogenicity. Analytical methods like liquid chromatography-mass spectrometry (LC-MS) and capillary electrophoresis (CE) are used to profile glycoforms.
5. Charge Variants and Isoform Characterization
- Protein drugs can exist in multiple isoforms due to charge variations arising from PTMs. Ion-exchange chromatography (IEX), capillary isoelectric focusing (cIEF), and MS are used to separate and characterize charge variants.
6. Functional Characterization
- Binding Assays: The binding affinity of antibodies to their target antigens is tested using surface plasmon resonance (SPR) or biolayer interferometry (BLI). This is critical for monoclonal antibodies to ensure they maintain their specific interaction with antigens.
- Bioactivity Assays: Cell-based assays or enzymatic activity assays (for enzyme-based protein drugs) are used to assess the biological activity of the drug.
7. Immunogenicity Assessment
- Immunogenicity is a major concern for protein-based drugs, as they can trigger an immune response. Structural characterization helps predict immunogenic potential by identifying aggregation, PTMs, and other structural anomalies.
8. Impurity and Degradation Product Analysis
- Impurities: These include host cell proteins, nucleic acids, or other contaminants from the manufacturing process. Techniques like enzyme-linked immunosorbent assay (ELISA) and MS are used for detection.
- Degradation Products: The identification of degradation pathways and products is essential for assessing the stability and shelf-life of the drug. Forced degradation studies combined with MS, HPLC, and SEC are typically employed.
9. Process-Related Variability and Batch Consistency
- During Phase III, ensuring batch-to-batch consistency is crucial. Analytical characterization is used to confirm that every batch produced during clinical trials has the same structural attributes and biological activity as the previous batches.
10. Regulatory and Quality Control Considerations
- Regulatory agencies like the FDA and EMA have specific guidelines (e.g., ICH Q6B) that detail the requirements for structural characterization of biologics. These include demonstrating the drug's identity, purity, potency, and safety.
Structural characterization of protein and antibody drugs during Phase III clinical trials is a multi-faceted process that involves a combination of analytical techniques to ensure product consistency, safety, and efficacy. This phase is critical for confirming that the biologic is suitable for regulatory approval and eventual commercial use. Accurate structural characterization ensures that the biologic will perform as intended in patients while minimizing the risk of adverse effects or loss of efficacy due to structural changes.










