Why Choose Creative Proteomics for Target Validation
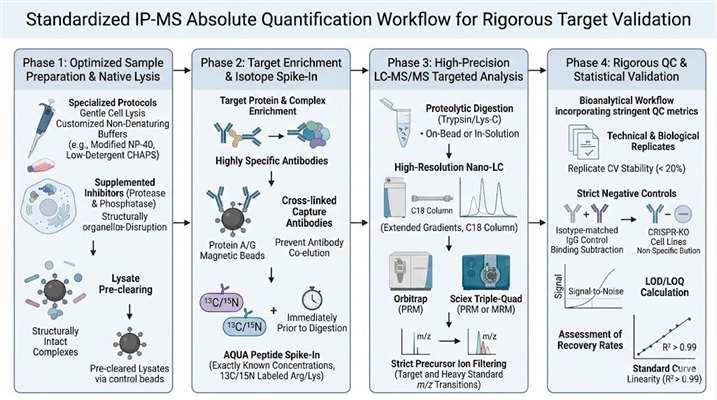
For high-stakes MoA research, sample loss, non-specific binding, and data ambiguity are critical roadblocks. Creative Proteomics brings extensive experience in specialized proteomics, addressing the unique pain points of top-tier academic labs and mid-to-large biopharma organizations.
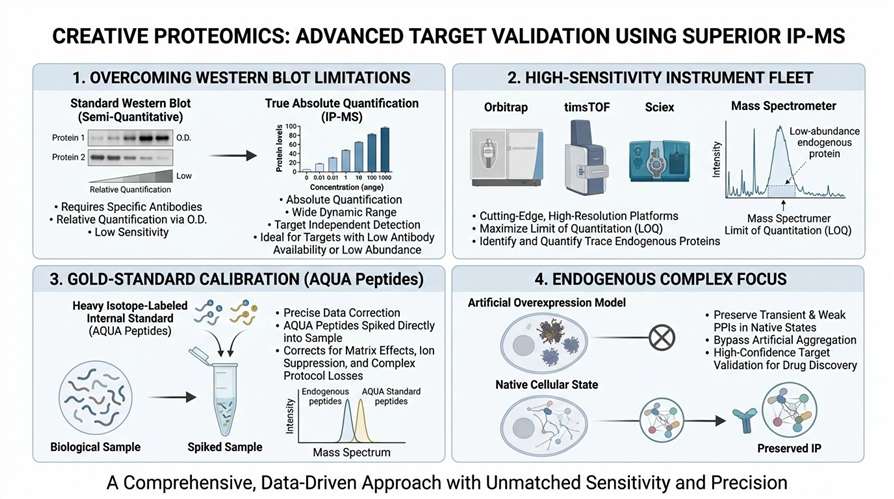
- Overcoming Western Blot Limitations: Eliminate reliance on semi-quantitative, antibody-dependent optical density. We provide true absolute quantification across a wide dynamic range, ideal for targets that fail standard detection methods due to low abundance or poor antibody availability for blotting.
- High-Sensitivity Instrument Fleet: All analyses are conducted on cutting-edge, high-resolution platforms, including Orbitrap, timsTOF, and Sciex systems, maximizing the limit of quantitation (LOQ) for trace endogenous proteins.
- Gold-Standard Calibration: We utilize heavy isotope-labeled internal standards (such as AQUA peptides) spiked directly into your samples, correcting for matrix effects, ion suppression, and sample loss during complex IP protocols.
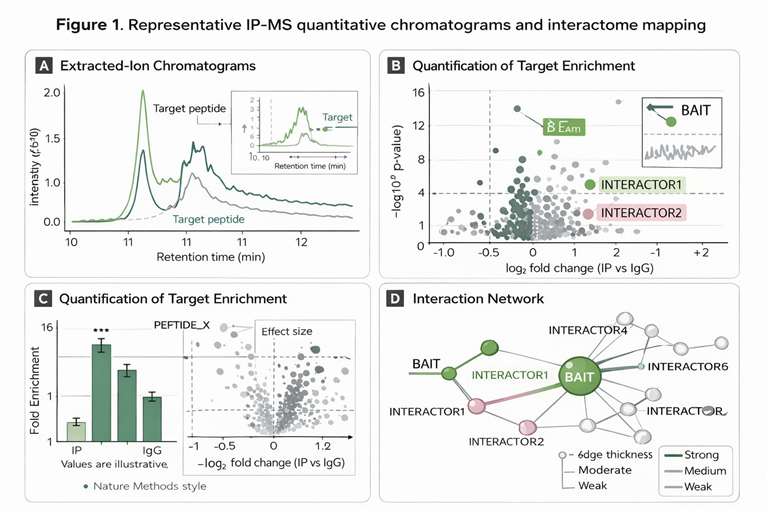
- Endogenous Complex Focus: Specialized protocols designed to preserve transient and weak protein-protein interactions (PPIs) in their native cellular states, bypassing the artificial aggregation often seen in overexpression models and delivering high-confidence target validation for drug discovery.