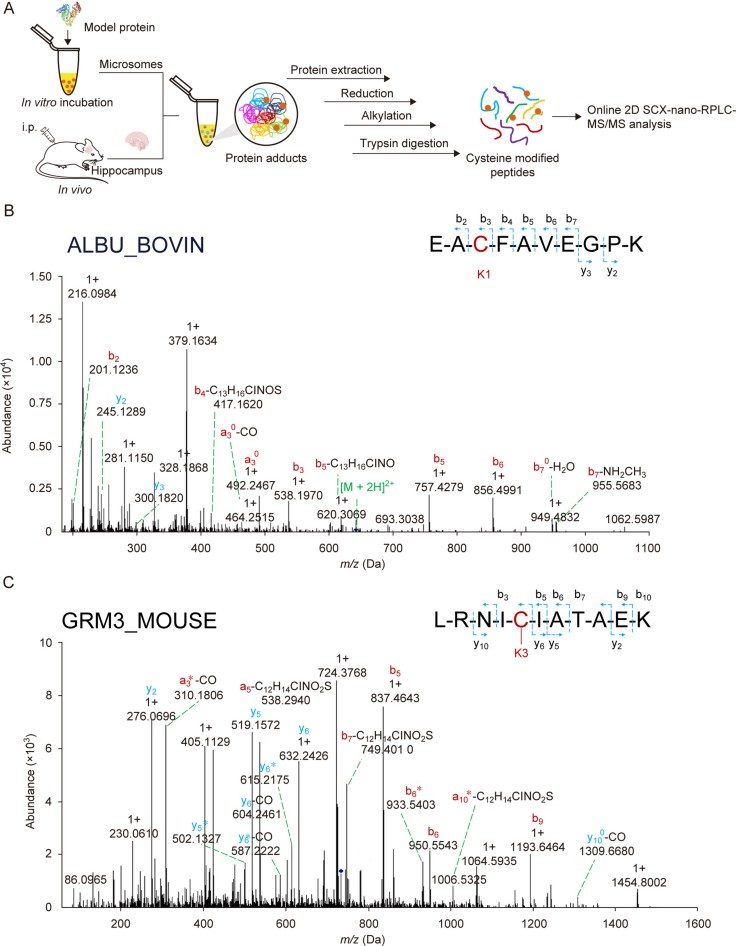
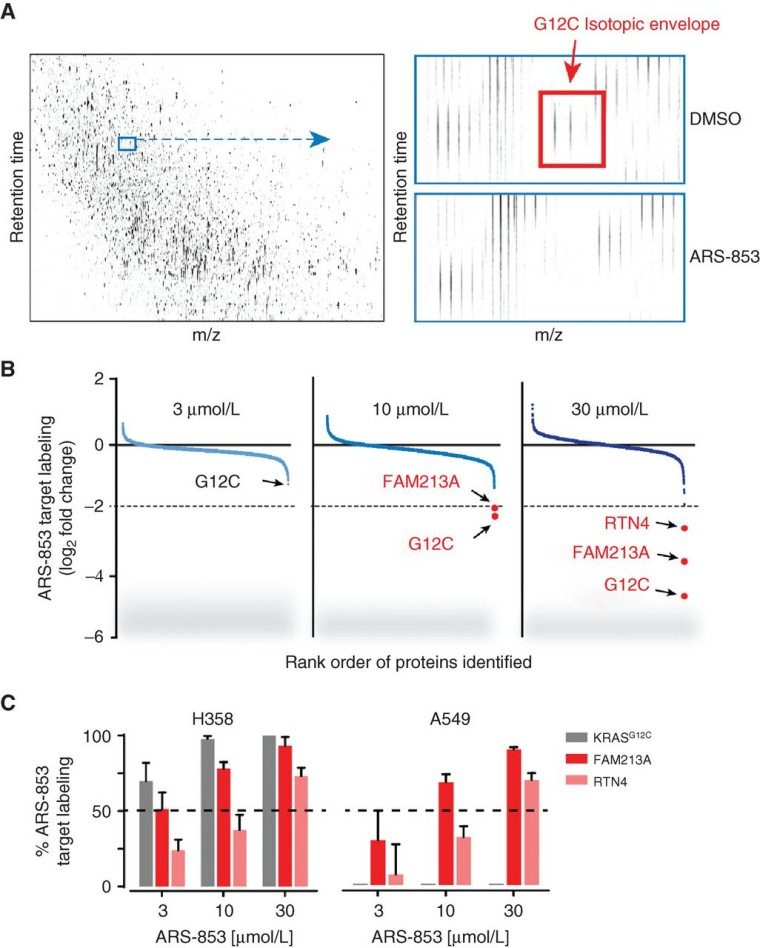
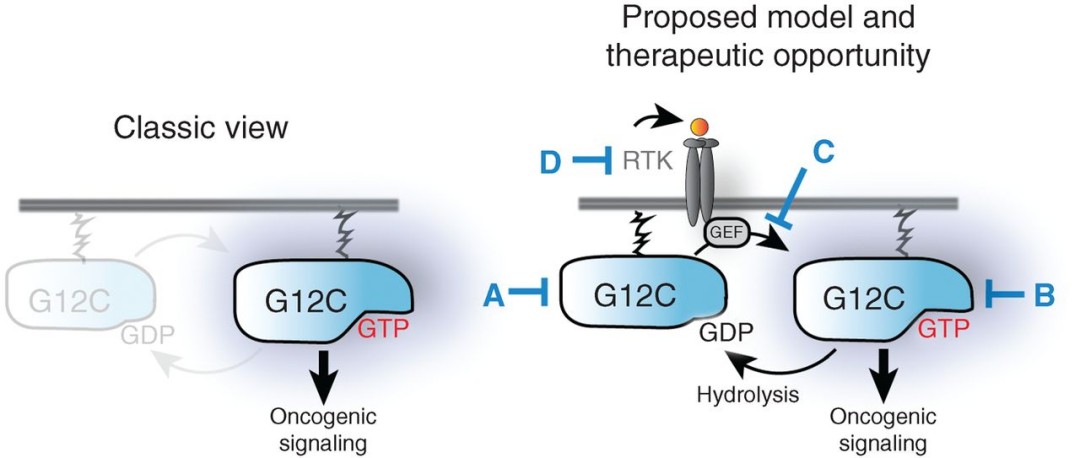
What is cysteine used for?
Covalent small molecules have gained renewed attention due to their ability to form stable, irreversible interactions with specific amino acid residues in proteins. Among these residues, cysteine stands out because of its unique chemical reactivity and relatively low abundance in the proteome. This combination enables selective targeting while minimizing widespread nonspecific binding.
From a proteomics perspective, understanding how small molecules interact with cysteine residues requires quantitative, site-resolved analytical strategies. Advances in mass spectrometry-based proteomics and chemoproteomics now allow systematic mapping of cysteine engagement across complex biological systems, providing critical insights into molecular selectivity and protein interaction landscapes.
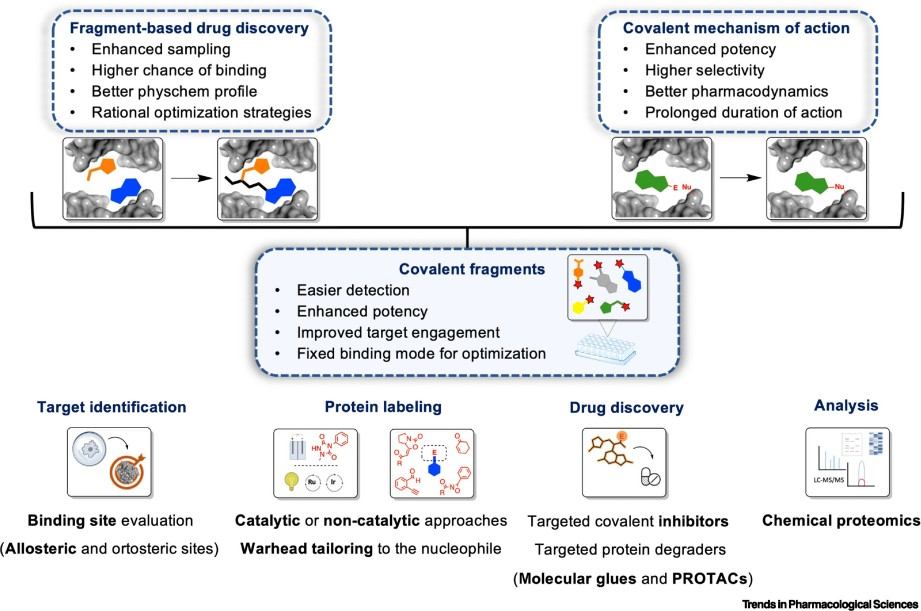
Figure 1. The concept of covalent fragments, their warheads and labeling chemistries (Csorba N, et al., 2023).
Chemical and Structural Characteristics of Cysteine
Cysteine contains a nucleophilic thiol (–SH) side chain that can participate in redox reactions, metal coordination, and covalent modification. In proteins, cysteine residues may be solvent-exposed, buried within folded structures, or involved in disulfide bonds, leading to diverse reactivity profiles.
This heterogeneity is a key reason why cysteine-targeted covalent interactions are highly context-dependent. Quantitative proteomics approaches are therefore essential to distinguish reactive, ligandable cysteines from those that are structurally inaccessible or functionally constrained.
Principles of Cysteine-Targeted Covalent Binding
Cysteine-targeted covalent molecules typically rely on electrophilic functional groups that react with thiol groups under physiological conditions. The binding process involves two conceptual steps:
- Recognition and positioning, where the molecule associates with the protein through non-covalent interactions
- Covalent bond formation, where the electrophile reacts with the cysteine thiol
Quantitative analysis of this process focuses on occupancy, selectivity, and competition at the residue level. These parameters cannot be reliably inferred from bulk assays alone and require proteome-wide measurement strategies.
Chemoproteomics Strategies for Cysteine-Targeted Covalent Drugs
Chemoproteomics integrates chemical probes with mass spectrometry to profile protein-small molecule interactions directly in complex samples. For cysteine-focused studies, thiol-reactive probes are commonly used to label accessible cysteine residues across the proteome.
When a covalent small molecule occupies a cysteine site, probe labeling at that position is reduced. By quantitatively comparing probe signals between treated and control samples, researchers can infer which cysteine residues are engaged and to what extent. This strategy enables unbiased identification of both primary interaction partners and secondary binding proteins.
Why Identifying Cysteine Binding Sites Is Critical
Knowing which cysteine residue is modified is as important as identifying the target protein itself. Site-specific information enables researchers to:
- Distinguish functional binding from nonspecific reactivity
- Compare binding patterns across compound analogs
- Support structure–activity relationship optimization
- Evaluate selectivity across protein families
Proteomics-based cysteine mapping provides this information directly at the peptide and residue level, offering clarity that cannot be achieved through bulk readouts alone.