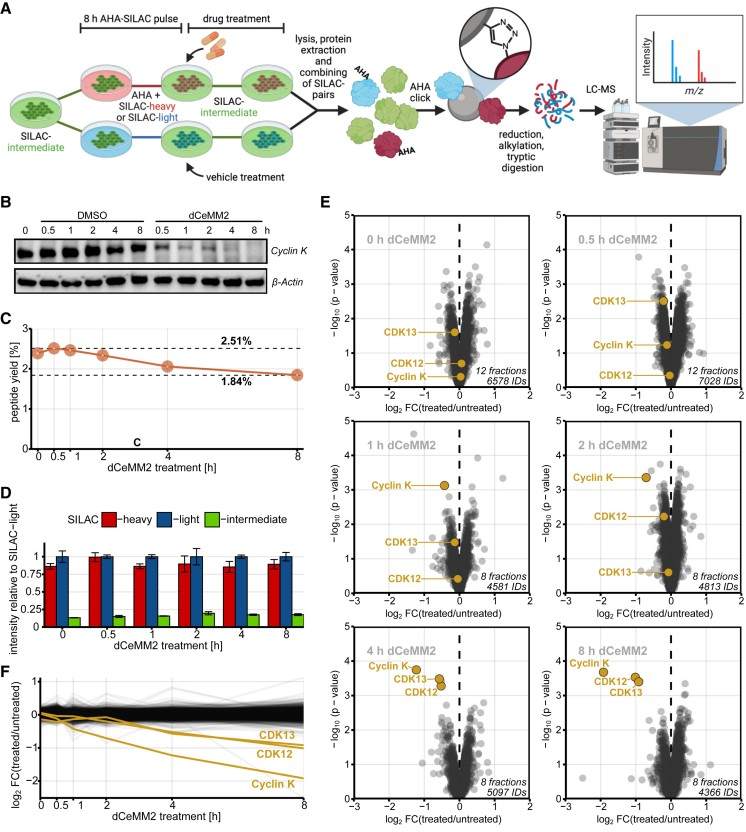
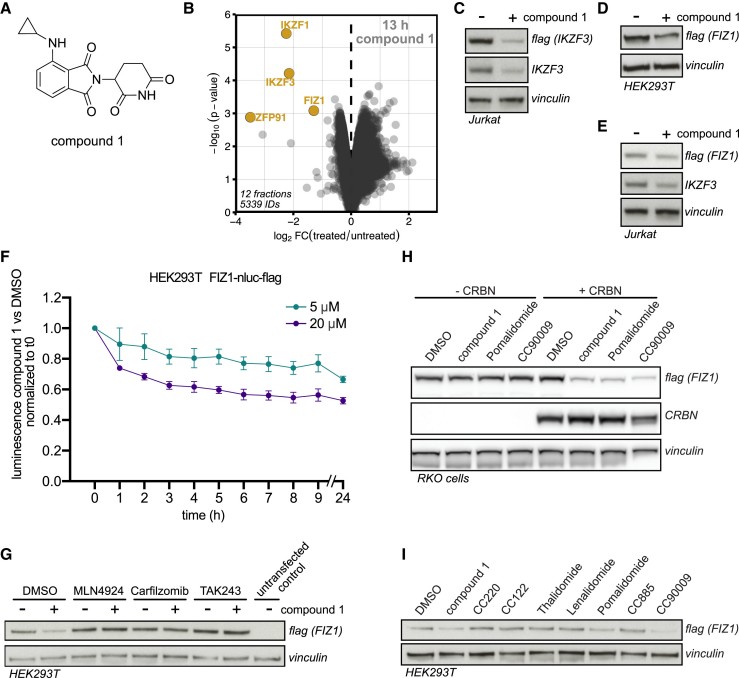
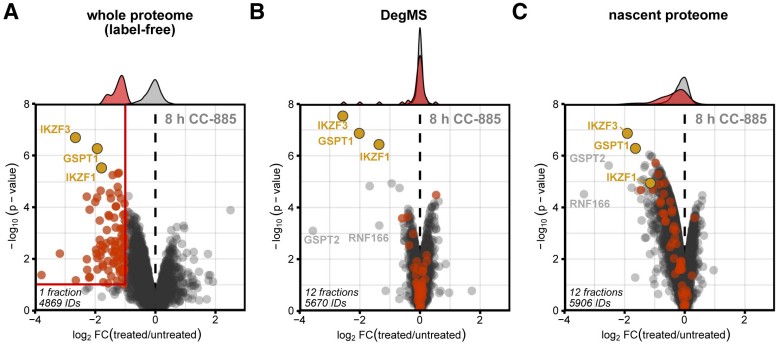
CORE SERVICE
We provide targeted protein degradation analysis services powered by quantitative proteomics and high-resolution mass spectrometry, helping researchers accurately measure degradation efficiency, selectivity, and proteome-wide impact. Our workflows are built on well-established proteomics methodologies, delivering reliable, reproducible, and interpretable results that support confident decision-making across degrader research projects.
- Quantitative degradation profiling to measure target protein loss across conditions and time points precisely
- Proteome-wide selectivity assessment to reveal unintended protein changes beyond the primary target
- High-confidence mass spectrometry analysis supported by robust quality control and statistical validation
- Expert-driven data interpretation grounded in extensive experience with complex proteomics datasets