Integrated Multi-Omics Strategy for High-Confidence Neoepitope Discovery
High-confidence neoepitope identification requires more than peptide detection alone. At Creative Proteomics, we integrate proteomics, genomics, and transcriptomics to ensure that each reported neoepitope is biologically real, tumor-specific, and relevant for downstream immunotherapy development.
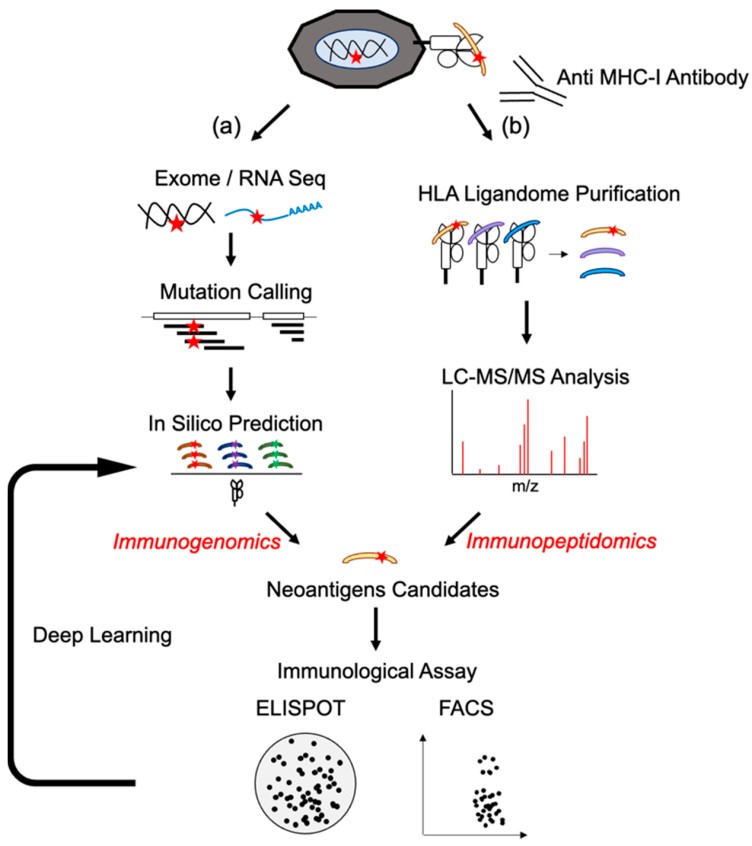
MS–based immunopeptidomics directly identifies peptides that are naturally displayed on tumor cell surfaces by MHC molecules. This confirms that a neoepitope is actually presented to the immune system.
Genomic analysis (such as whole-exome or whole-genome sequencing) links each identified peptide to its underlying DNA mutation. This step verifies that the neoepitope originates from a tumor-specific genetic change and is absent from normal tissues.
Transcriptomic data (RNA sequencing) adds a third layer of confidence by confirming that the mutated gene is actively expressed in the tumor. Neoepitopes derived from genes that are not expressed are unlikely to be presented at meaningful levels and are therefore deprioritized.
Bioinformatics Pipeline: From Raw Spectra to Prioritized Neoepitopes
Key Analytical Steps:
- Somatic mutation calling and annotation
- Customized proteogenomic database construction
- Peptide spectrum matching with stringent FDR control
- HLA typing and MHC binding affinity prediction
- Neoepitope prioritization based on presentation confidence, expression, and immunogenic potential
Advantages of a Proteomics-Driven Neoepitope Strategy
Compared with sequence-based prediction alone, proteomics-driven neoepitope identification offers:
- Experimental confirmation of peptide presentation
- Reduced false-positive candidate lists
- Access to non-canonical antigen sources
- Improved biological relevance for downstream research
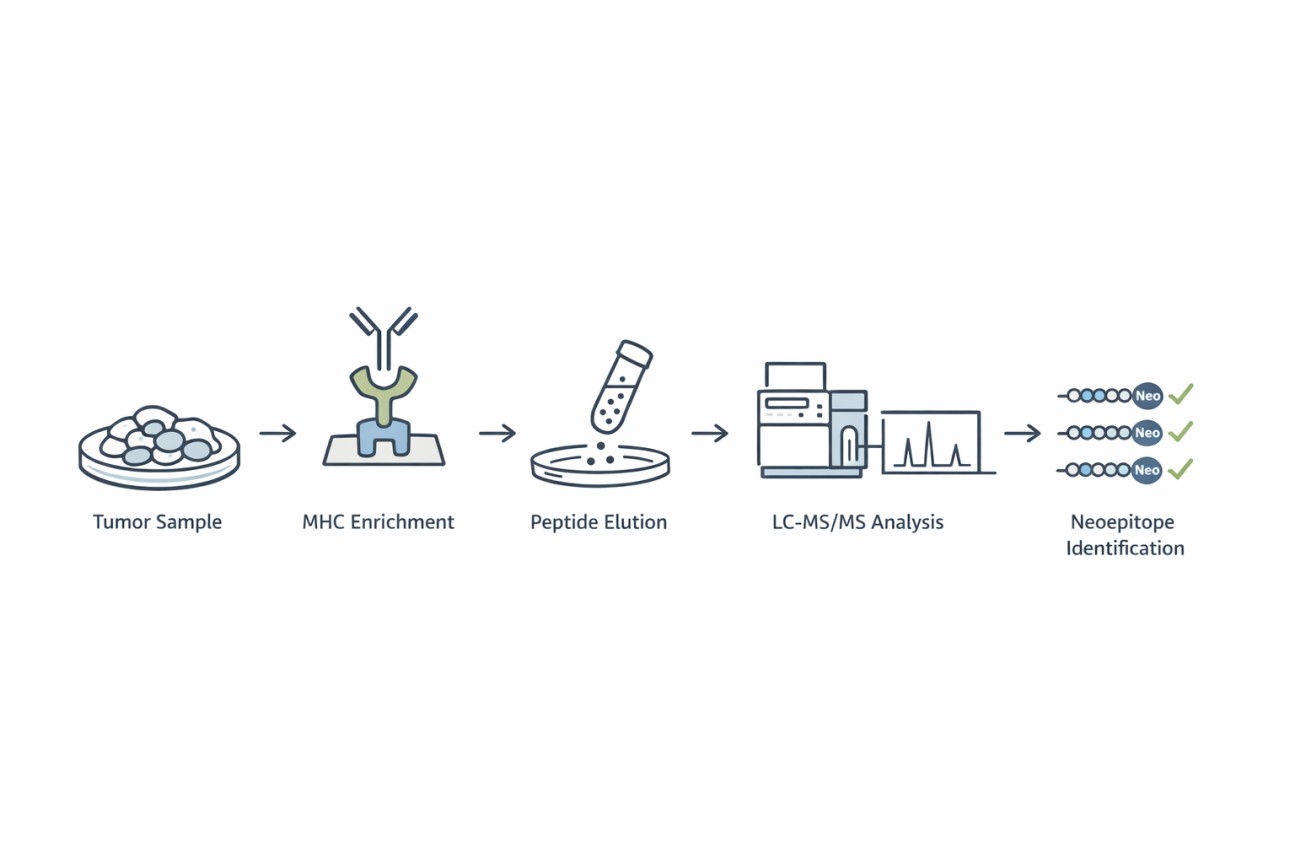
Creative Proteomics' Neoepitope Identification Service Workflow
- MHC Enrichment: High-affinity monoclonal antibodies are used to isolate MHC class I and/or class II complexes from tumor samples.
- Peptide Elution: MHC-bound peptides are gently eluted without disrupting peptide integrity.
- LC-MS/MS Analysis: High-resolution mass spectrometry identifies peptide sequences with high sensitivity and accuracy.
- Bioinformatic Annotation: Identified peptides are matched against customized proteogenomic databases to distinguish neoepitopes from self-peptides.
Deliverables: What You Receive
Clients receive a comprehensive and actionable dataset, including:
- Identified MHC-bound peptide lists with MS evidence
- Annotated variant files (VCF/TSV)
- HLA typing results
- Prioritized neoepitope candidates
- Summary reports with interpretation guidance
Validation Strategies and Downstream Support
To further increase confidence, Creative Proteomics offers optional validation strategies:
Applications in Cancer and Immunology Research
- Studying antigen processing and presentation mechanisms
- Investigating tumor immune evasion strategies
- Comparing neoepitope landscapes across tumor subtypes
- Supporting exploratory immune profiling studies