What Is the Difference Between CDX and PDX Models?
| Feature |
PDX Models |
CDX Models |
| Source of Tumor Material |
Established by directly implanting tumor tissue from a patient into immunodeficient or humanized mice. |
Derived from commercially available or in-house cultured tumor cell lines. |
| Biological Fidelity |
High fidelity to the original human tumor. Maintains histopathology, genetic heterogeneity, tumor microenvironment characteristics, and signaling networks. |
Moderate biological fidelity. Tumor cell lines are often more homogeneous and may have adapted to in vitro culture conditions. |
| Experimental Throughput |
Lower throughput due to limited availability of patient tissue and slower tumor engraftment. |
High throughput. Cell lines are readily expandable and allow rapid establishment of multiple parallel experiments. |
| Tumor Heterogeneity |
Captures intra- and inter-patient heterogeneity, enabling study of diverse molecular subtypes and resistance mechanisms. |
Less heterogeneous. CDX models generally represent a single clone, providing more controlled and reproducible experimental conditions. |
| Applications |
Optimal for translational studies, biomarker identification, and validation of therapeutic strategies in clinically relevant models. |
Well-suited for drug screening, target validation, pharmacokinetics/pharmacodynamics (PK/PD) studies, and rapid evaluation of multiple compounds or treatment regimens. |
| Time and Cost Considerations |
Longer engraftment times and higher costs due to tissue acquisition and animal maintenance. |
Faster to establish and lower cost, making CDX models practical for larger-scale studies and early-stage preclinical evaluation. |
| Integration with Proteomics |
Provides highly relevant protein expression and pathway profiling reflecting patient-specific tumor biology, supporting translational and preclinical assessment. |
Supports reproducible proteomic profiling across multiple experiments, enabling comparative studies and validation of targets or pathways identified in PDX models. |
Quantitative Proteomics Strategies for PDX/CDX Studies
Quantitative proteomics is a core experimental approach for characterizing PDX and CDX animal models at the protein level. It enables systematic measurement of protein abundance changes within tumor tissues, providing a direct view of molecular processes operating in vivo. In PDX/CDX studies, quantitative proteomics is commonly used to investigate treatment-induced effects, compare tumor subtypes, and explore biological differences across models under controlled experimental conditions.
Label-Free Quantitative Proteomics
Label-free quantification relies on repeated measurement of peptide signal intensities or spectral counts across multiple mass spectrometry runs. In PDX/CDX studies, it is frequently employed when analyzing large numbers of xenograft tumors or when sample availability varies across different models.
Sample preparation, chromatographic conditions, and instrument parameters are tightly controlled to ensure that differences in signal reflect biological variation rather than technical noise. Quantification is typically performed using intensity-based algorithms that align peptide features across runs and calculate relative protein abundance across experimental groups.
Label-free workflows are well suited for discovery-oriented studies:
- Mapping global protein expression patterns.
- Identifying pathways altered by compound exposure.
- Comparing baseline proteomic profiles across different PDX or CDX models.
Label-Based Quantitative Proteomics
Label-based strategies enable the simultaneous analysis of multiple samples in a single mass spectrometry run. Each sample is assigned a chemical tag, enabling precise relative comparison across experimental groups. In PDX/CDX experiments, label-based strategies are often used when an accurate, direct comparison between defined conditions is required. Tumor samples are processed in parallel, labeled individually, pooled, and analyzed together to reduce experimental variability.
This strategy is particularly useful for:
- Direct comparison across treatment arms is required.
- Subtle protein changes need to be measured with high confidence.
- Batch-to-batch variability must be minimized.
Creative Proteomics' Label-based quantitative proteomics services:
DIA-Based Proteomics for PDX/CDX Profiling
Data-independent acquisition (DIA) has become a core strategy for proteomic analysis of PDX and CDX animal models because it delivers consistent, reproducible, and comprehensive protein measurements across complex in vivo samples. Unlike traditional data-dependent methods that selectively analyze only the most abundant peptides, DIA systematically fragments all detectable peptides within defined mass ranges, ensuring that biologically relevant proteins are not missed due to sampling bias.
Why DIA is Well-Suited for PDX/CDX Studies
PDX and CDX tumor tissues are inherently heterogeneous and often available in limited amounts. DIA addresses these challenges by providing:
- High data completeness, allowing the same proteins to be quantified across all models and treatment groups.
- Excellent run-to-run reproducibility, which is essential for comparing multiple animal cohorts.
- Stable quantification of low-abundance signaling proteins, including kinases and pathway regulators that drive tumor behavior.
Improved Throughput Without Sacrificing Biological Depth
Modern DIA workflows enable high sample throughput while maintaining deep proteome coverage. This allows large PDX or CDX panels to be analyzed within practical timelines, supporting:
- Head-to-head comparison of multiple tumor models
- Longitudinal studies tracking molecular changes over time
- Efficient screening of treatment and control groups
Reliable Quantification for Translational Research
Because DIA generates highly consistent quantitative data, it is well suited for downstream biological interpretation. Protein-level changes can be confidently linked to altered cellular pathways, enabling clearer insights into mechanism of action, pathway activation, and potential biomarker candidates. When combined with genomic or transcriptomic data, DIA-based proteomics strengthens the functional understanding of PDX and CDX models in translational research settings.
Applications of PDX/CDX Proteomic Characterization
Biomarker Discovery for Translational Research
Proteomic analysis of PDX and CDX models enables the systematic identification of proteins whose abundance or activity changes in response to disease progression or therapeutic intervention.
Mechanism-of-Action and Target Validation
Proteomic characterization provides direct evidence of how a test compound influences cellular pathways within PDX and CDX tumors. Measuring changes in protein abundance and signaling activity allows researchers to confirm whether a compound is affecting its intended biological target.
Model Selection and Biological Relevance Assessment
Not all PDX or CDX models are equally representative of human disease. Proteomic profiling allows objective comparison of models based on their functional molecular features rather than histology alone.
Therapy Response and Resistance Analysis
Quantitative proteomics enables detailed comparison of responding and non-responding PDX/CDX tumors following treatment. These analyses can uncover protein changes associated with reduced sensitivity, adaptive signaling, or survival mechanisms.
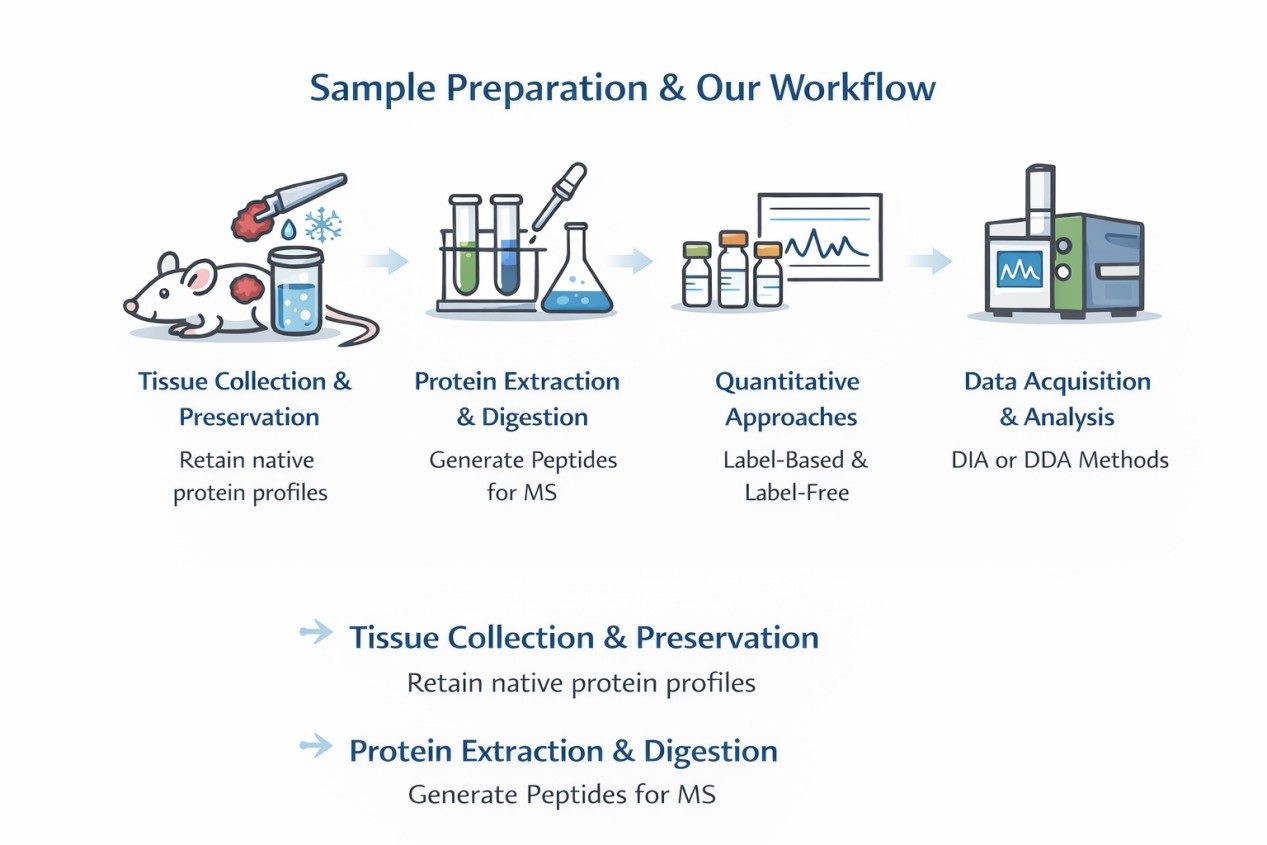
Sample Preparation and Our Workflow
- Tissue Collection and Preservation: Tumor fragments are collected from xenografted mice and preserved to retain native protein profiles.
- Protein Extraction and Digestion: High-efficiency lysis and enzymatic digestion generate peptides suitable for high-resolution mass spectrometry.
- Quantitative Approaches: Both label-based (TMT, iTRAQ) and label-free methods are applied, depending on study design.
- Data Acquisition and Analysis: DIA or DDA acquisition modes on Orbitrap or timsTOF platforms provide comprehensive proteome coverage.