CORE SERVICE
What is precise quantification of human energy metabolism enzymes?
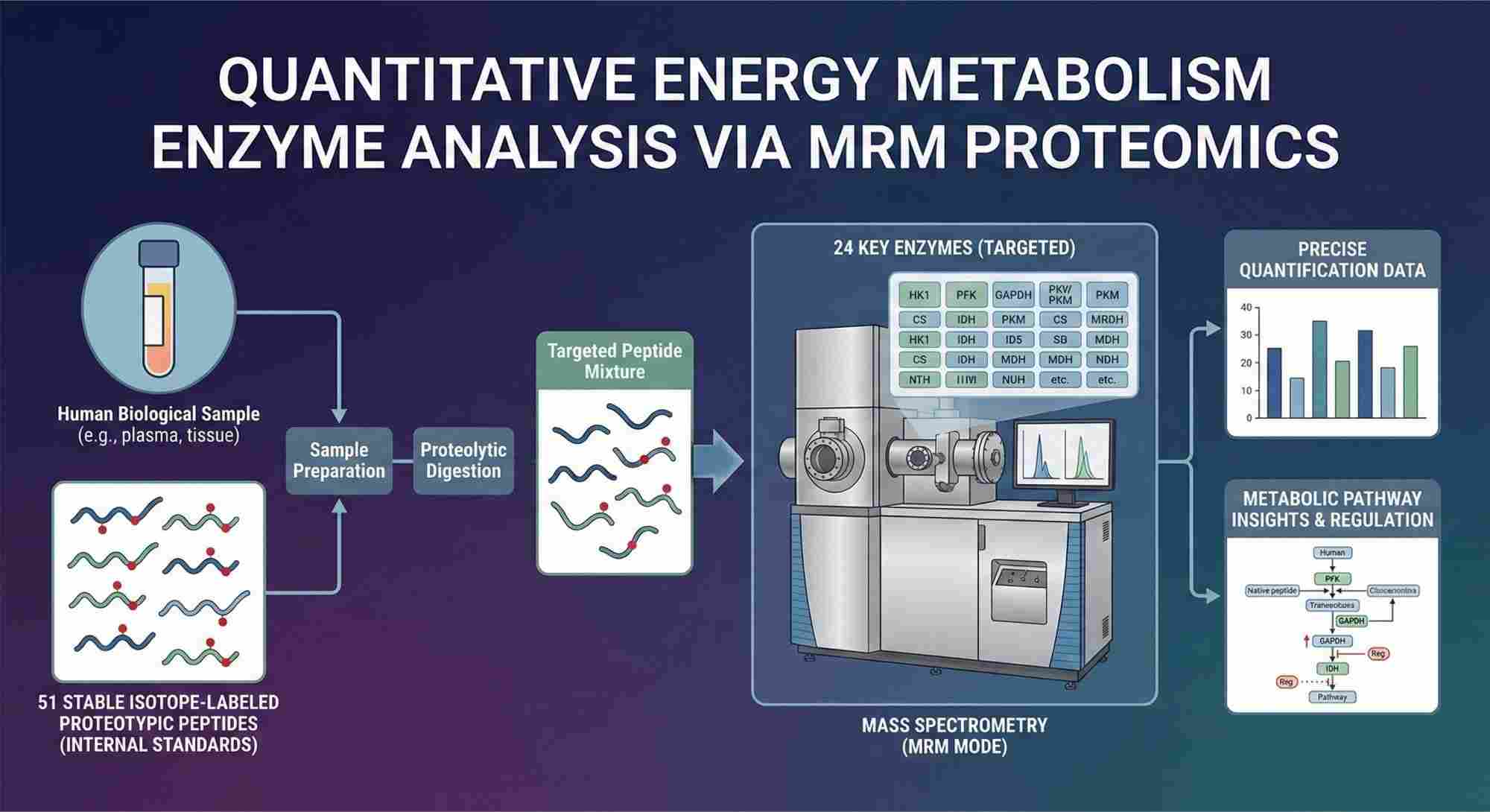
The precise quantification of human energy metabolism enzymes is achieved through a high-stringency targeted proteomics workflow utilizing Multiple Reaction Monitoring (MRM) mass spectrometry. This analytical framework relies on the principle of stable isotope dilution, wherein 51 distinct, stable isotope-labeled proteotypic peptides are employed as internal standards to target 24 specific enzymes. By introducing these heavy-labeled standards—which are chemically identical to the endogenous peptides but distinguishable by mass—into the biological matrix, the methodology rigorously corrects for ionization suppression, matrix effects, and sample processing variability. This ensures that the mass spectral signal intensity correlates directly and linearly to the absolute abundance of the target proteins, offering a level of accuracy superior to traditional relative quantification methods.
From a functional biology perspective, this approach facilitates the stoichiometric mapping of the central energy metabolism network, encompassing glycolysis, the tricarboxylic acid (TCA) cycle, and oxidative phosphorylation. Determining the absolute molar concentrations of these rate-limiting enzymes allows for the construction of accurate metabolic flux models and the assessment of bioenergetic capacity. This quantitative depth is essential for elucidating the kinetic constraints of cellular metabolism, enabling the precise characterization of metabolic phenotypes and the identification of regulatory bottlenecks underlying complex human biological states and disease pathologies.