CORE SERVICE
Biomarker Validation by PRM/MRM — Multiplexed Targeted Quantification for Biomarker Verification
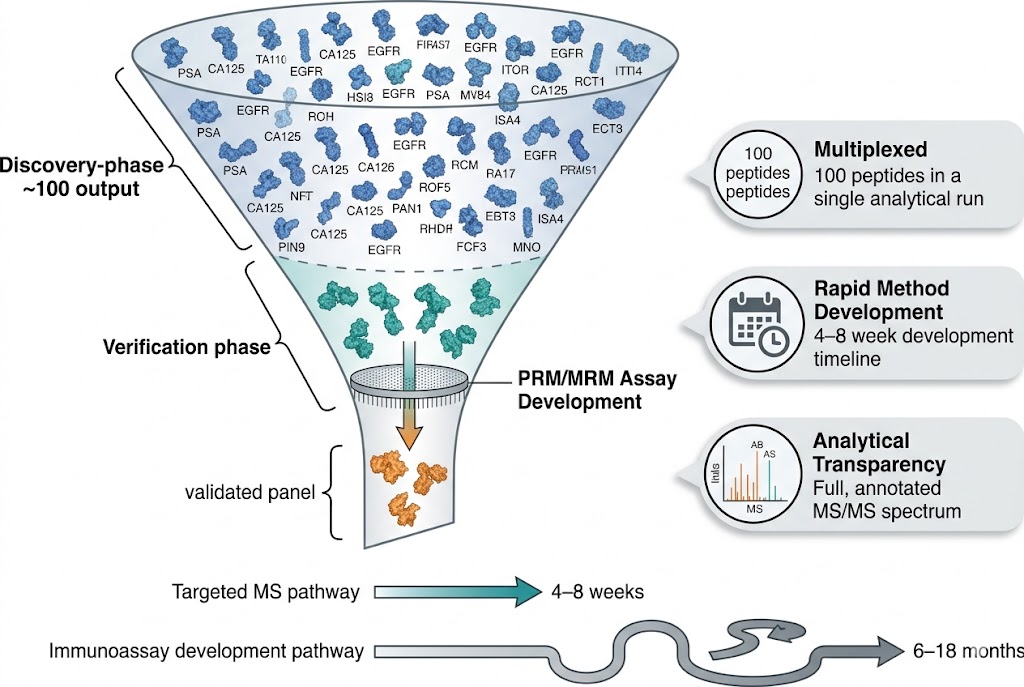
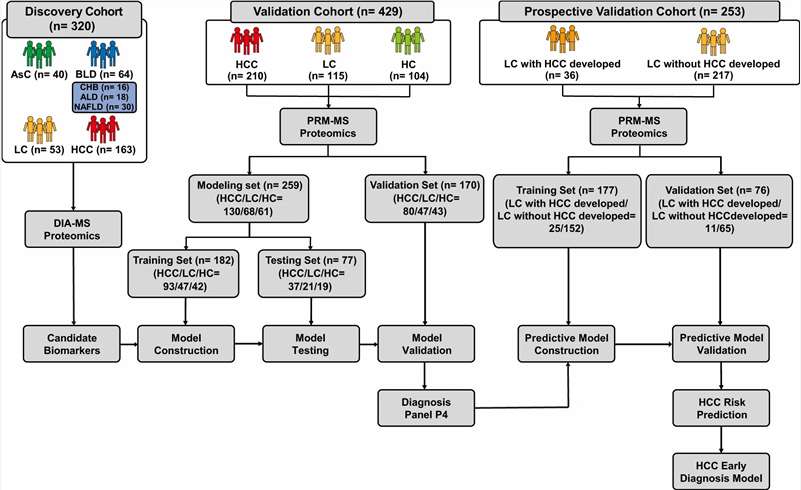
The transition from discovery-phase biomarker identification to robust, analytically validated assays is the most critical bottleneck in the biomarker development pipeline. Discovery proteomics (DIA, TMT, label-free) routinely generates hundreds of candidate biomarkers, but converting these candidates into quantitatively reliable, multiplexed assays suitable for cohort-scale validation requires a fundamentally different analytical approach. Immunoassay-based validation is constrained by antibody availability, cross-reactivity, and limited multiplexing capacity — typically 1–10 targets per assay.
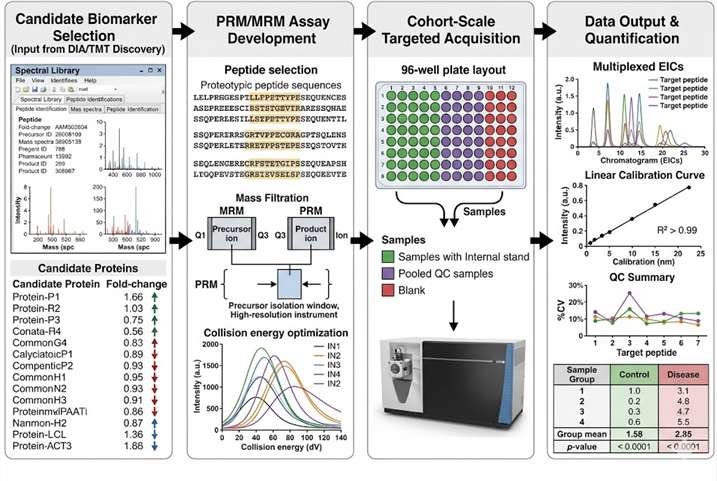
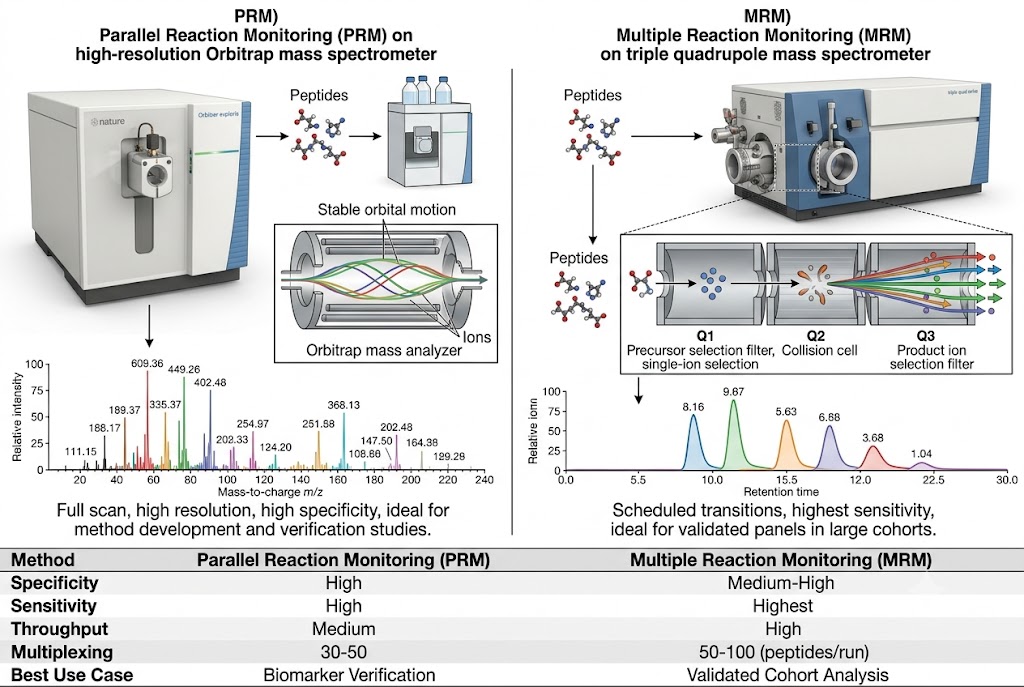
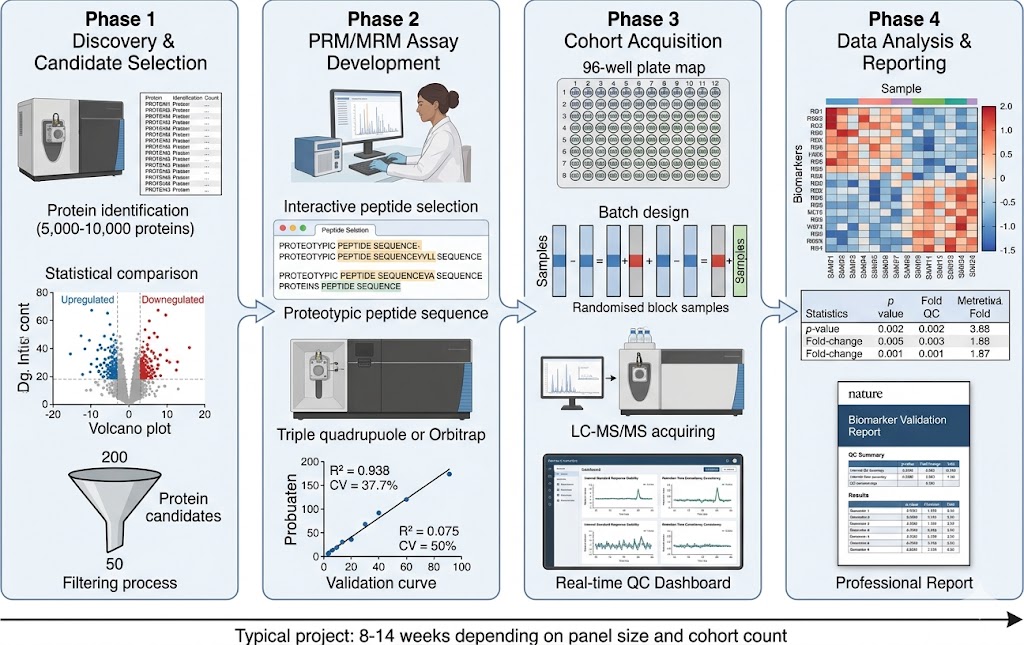
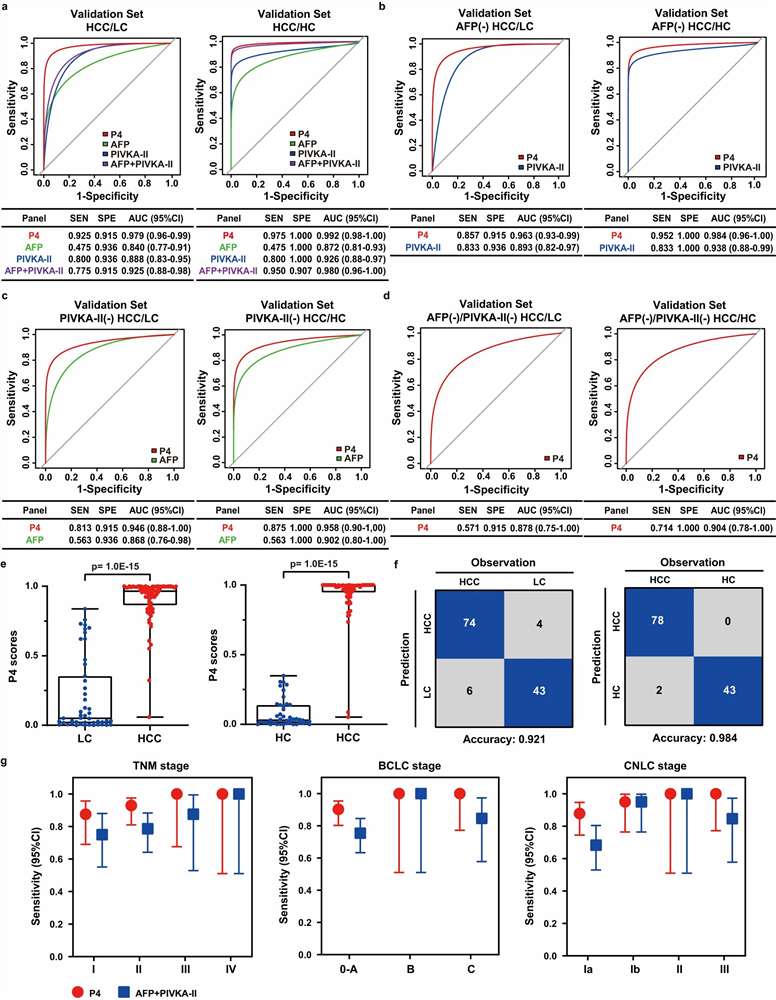
Our Biomarker Validation by PRM/MRM service addresses this gap directly. Using targeted LC-MS/MS acquisition on high-resolution Orbitrap/QqTOF (PRM) and triple quadrupole (MRM) platforms, we develop custom multiplexed assays that quantify 10–100+ candidate biomarkers simultaneously in a single analytical run. Stable isotope-labeled internal standards (AQUA/SIS peptides) provide absolute or relative quantification with documented analytical performance, while batch-randomised study designs and systematic QC frameworks ensure data quality across large cohorts.
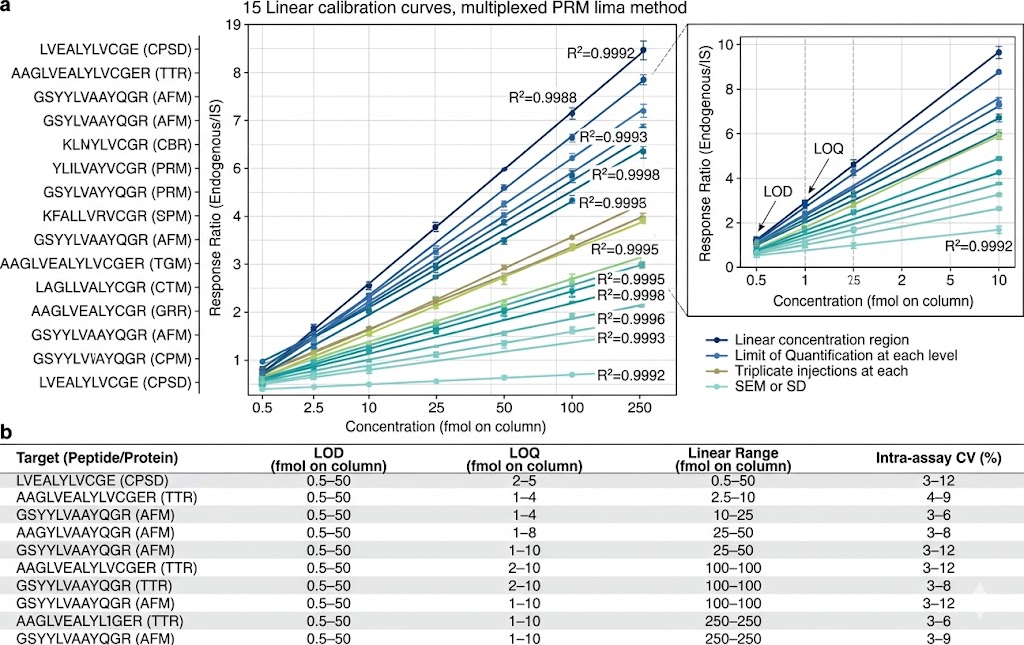
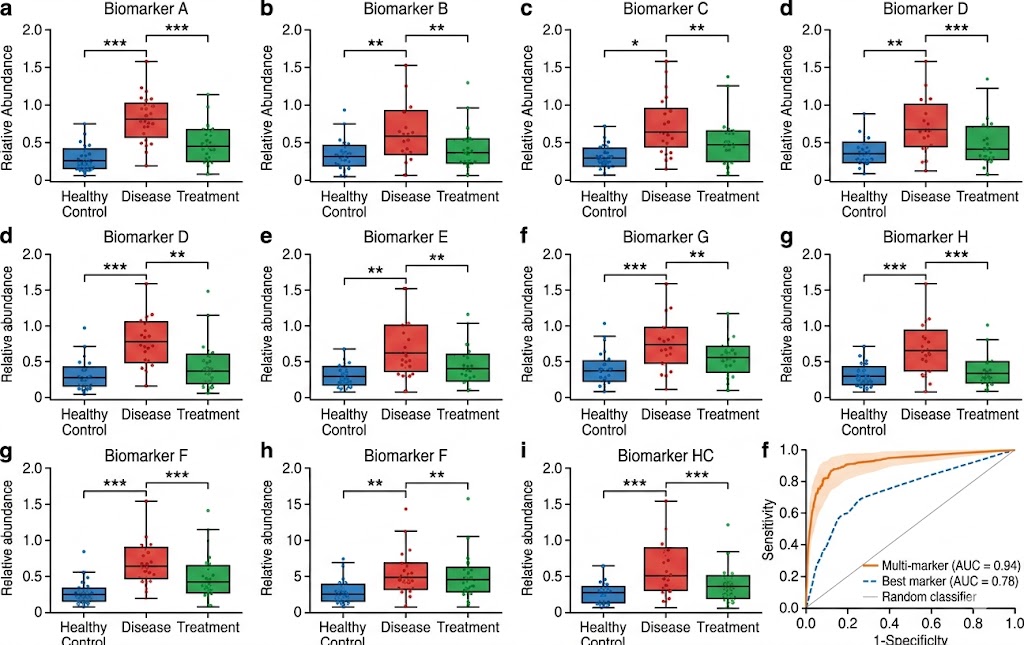
- Multiplexed Targeted Quantification: 10–100+ candidate biomarkers quantified simultaneously in a single scheduled LC-MS/MS run using PRM or MRM acquisition. Stable isotope-labeled internal standards for each target enable absolute quantification (fmol/µg protein) or standardised relative quantification with defined LOD, LOQ, linear dynamic range, and intra-/inter-assay CV metrics.
- Discovery-to-Validation Pipeline: Seamless transition from DIA/TMT discovery data to PRM/MRM validation assays. Our team designs targeted methods directly from discovery spectral libraries, selects proteotypic peptides with optimal MS properties, optimises transitions and collision energies, and applies the validated method to full cohort analysis — preserving biological context from discovery through validation.
- Cohort-Scale Reproducibility: Batch-randomised study designs with pooled QC injections every 5–10 samples, internal standard response monitoring, blank injections, and inter-batch CV assessment. Our reporting package documents all QC metrics, enabling reviewers and collaborators to assess data quality independently.