CORE SERVICE
End-to-End Plasma & Serum Biomarker Discovery: From DIA Profiling to PRM-Validated Panels
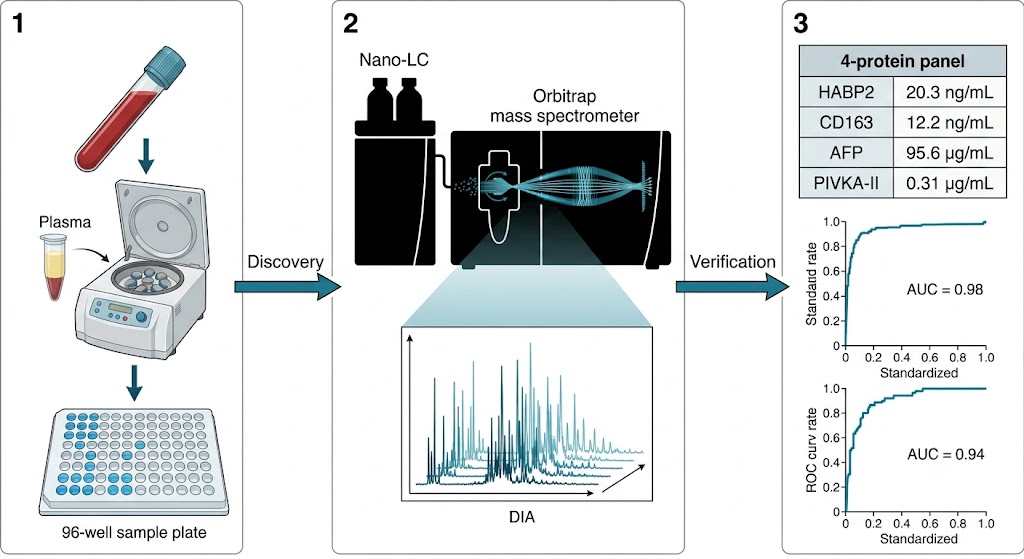
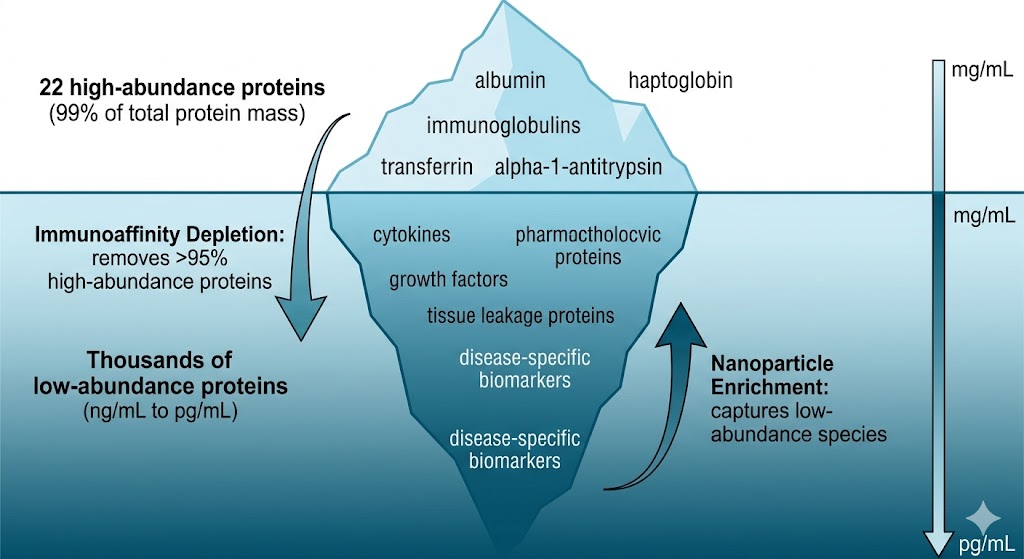
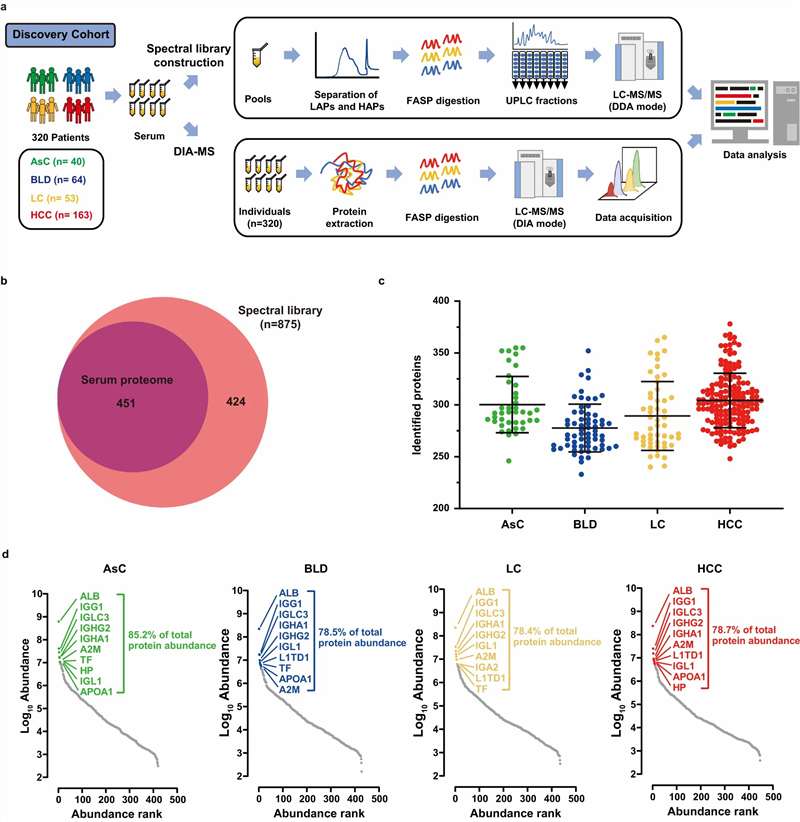
The path from a banked plasma cohort to a validated protein biomarker panel is fragmented across most CROs — discovery goes to one lab, targeted verification to another, and statistical panel optimization falls through the cracks entirely. Plasma and serum are the most accessible and clinically relevant sample types for protein biomarker research, yet they are also the most analytically demanding: 22 high-abundance proteins account for 99% of total protein mass, burying thousands of low-abundance biomarker candidates beneath a dynamic range spanning ten orders of magnitude. Our Plasma & Serum Biomarker Proteomics service closes this fragmentation by delivering the entire pipeline — DIA discovery, PRM/MRM verification, and machine learning-driven panel selection — as a single integrated workflow. Whether your study targets oncology biomarkers in pre-treatment serum, neurodegenerative protein signatures in plasma, or inflammatory markers across a longitudinal cohort, our Biomarker Validation by PRM/MRM platform ensures that candidates identified in discovery move seamlessly into statistically powered verification without the data loss and methodological drift that plague multi-vendor biomarker programs.
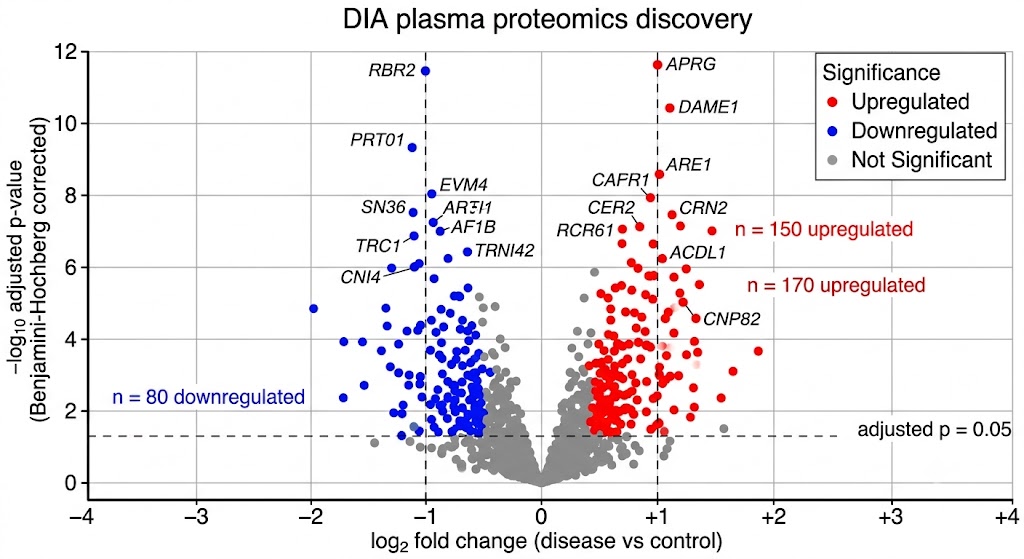
- Deep DIA Discovery Profiling: Data-independent acquisition on high-resolution Orbitrap and timsTOF platforms identifies and quantifies 800–1,500+ proteins per plasma sample after depletion, with systematic precursor fragmentation that creates a complete digital record free from the stochastic sampling bias of data-dependent acquisition. Every peptide in every sample is recorded — enabling retrospective re-analysis when new biomarker hypotheses emerge.
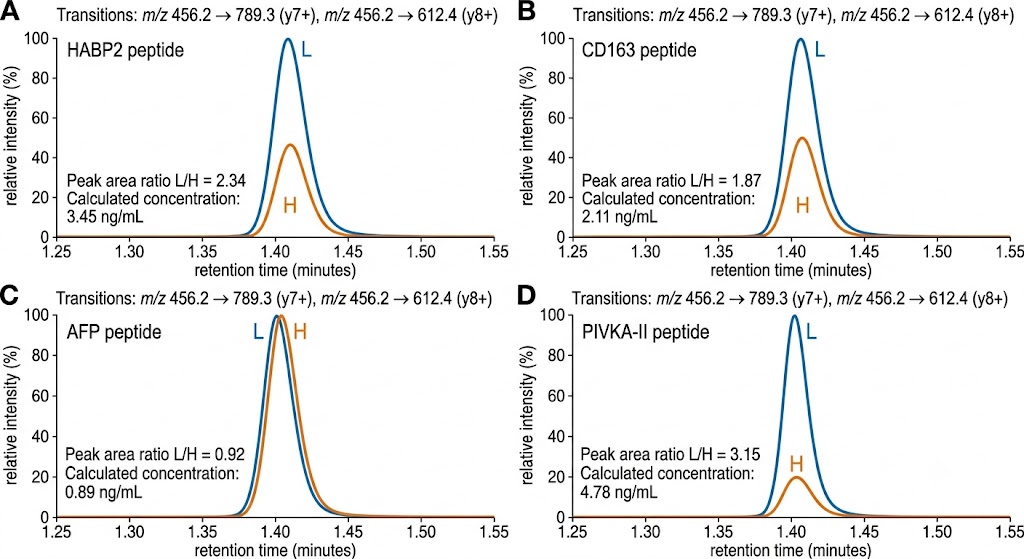
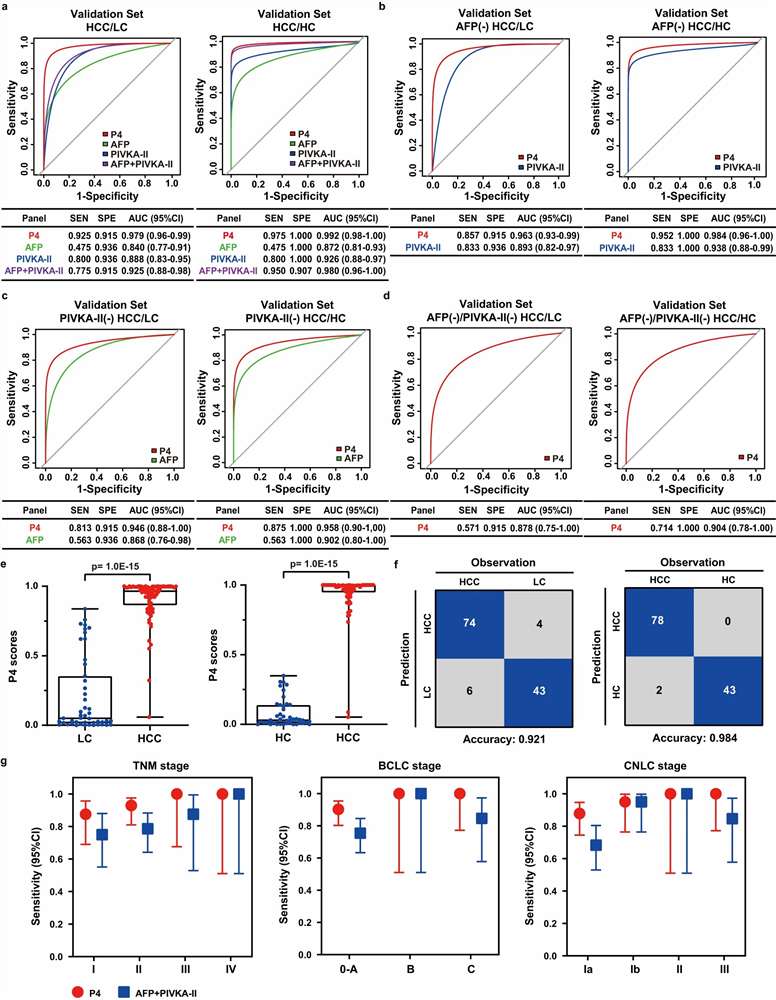
- PRM/MRM Targeted Verification: Custom assay development for 10–50 prioritized biomarker candidates, selecting 2–3 proteotypic peptides per protein with heavy isotope-labeled internal standards. Scheduled retention time windows and optimized collision energies deliver CVs below 15% across analytical replicates, with LODs reaching low ng/mL in depleted plasma — the quantitative rigor needed to separate true biomarker signal from cohort noise.
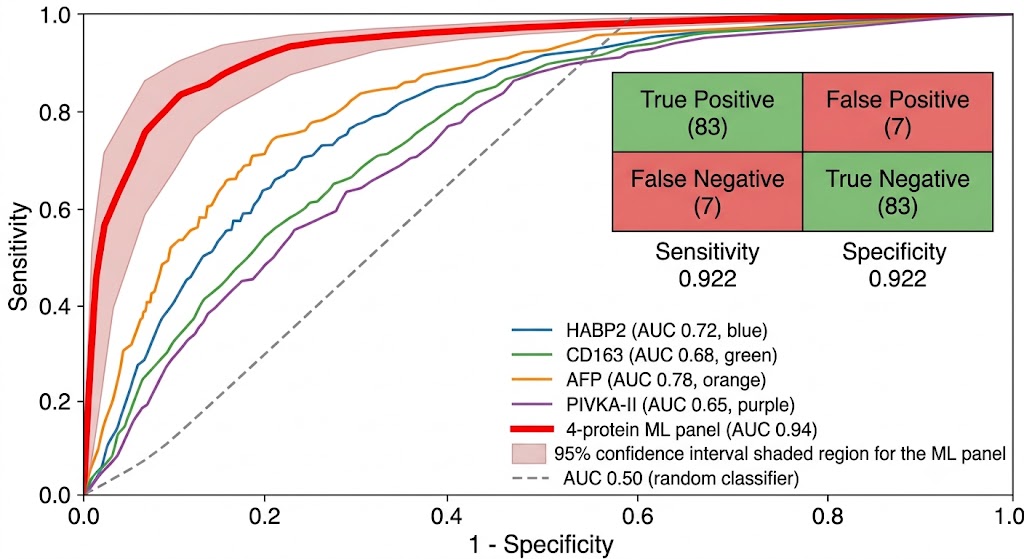
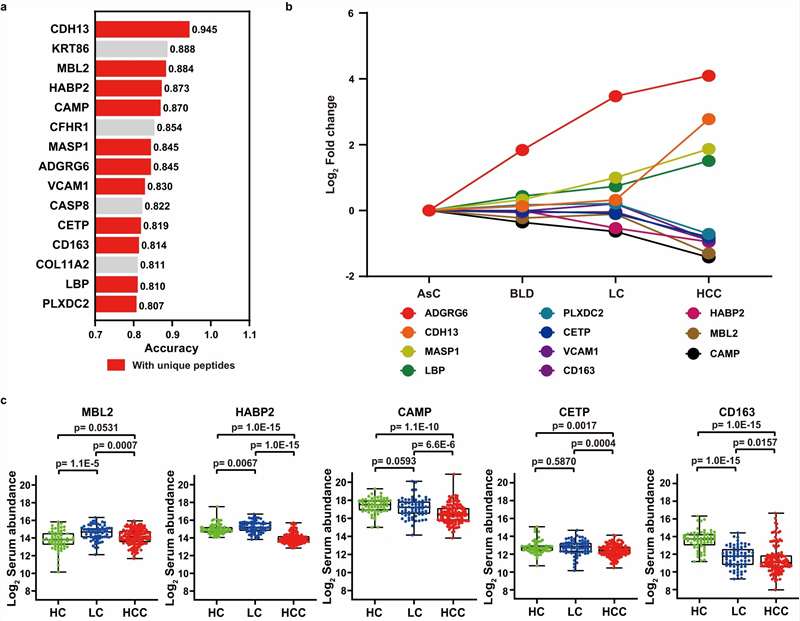
- Machine Learning Panel Optimization: Beyond simple fold-change ranking, we apply LASSO regression, random forest feature selection, and logistic regression modeling to identify the minimal protein panel that maximizes classification performance. Cross-validation and bootstrapping prevent overfitting, and the final panel is evaluated on held-out validation cohorts with ROC analysis, sensitivity/specificity metrics, and calibration curves.