CORE SERVICE
Integrated Proteome-Wide Mechanism of Action Profiling
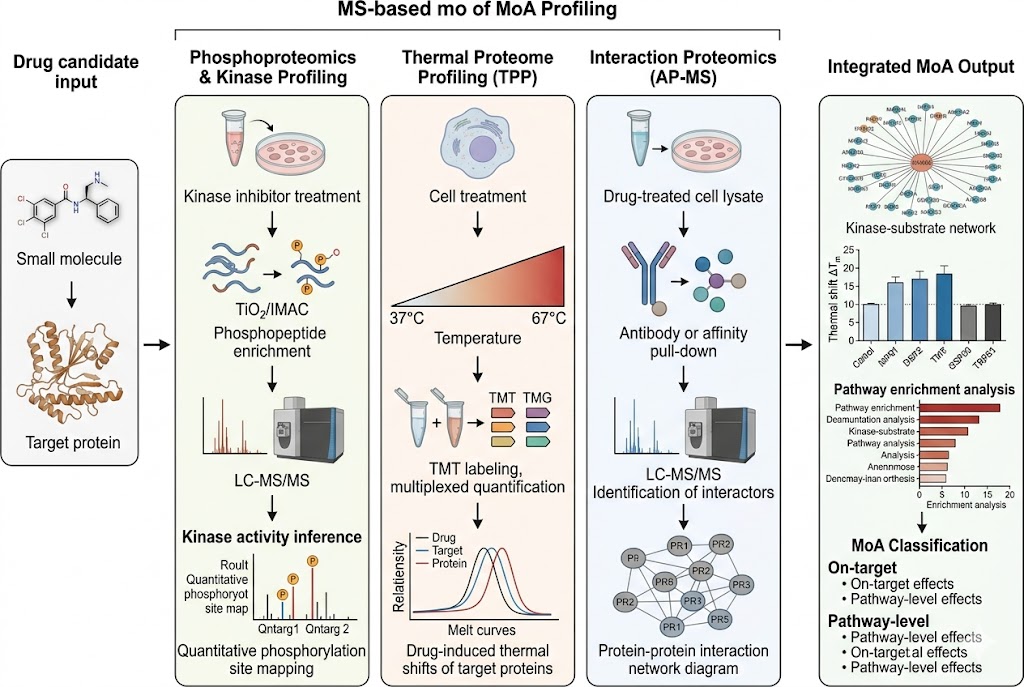
Understanding precisely how a drug candidate engages its target, modulates cellular signalling networks, and affects the wider proteome is fundamental to successful drug discovery. Traditional single-target assays provide limited mechanistic depth, often missing compensatory pathway activation, off-target pharmacology, or polypharmacology that may determine preclinical advancement outcomes. Our MoA Profiling service deploys three complementary mass spectrometry-based platforms — phosphoproteomics and kinase activity profiling, thermal proteome profiling (TPP), and interaction proteomics (AP-MS) — to deliver a comprehensive, systems-level view of drug mechanism from direct target engagement through downstream pathway effects.
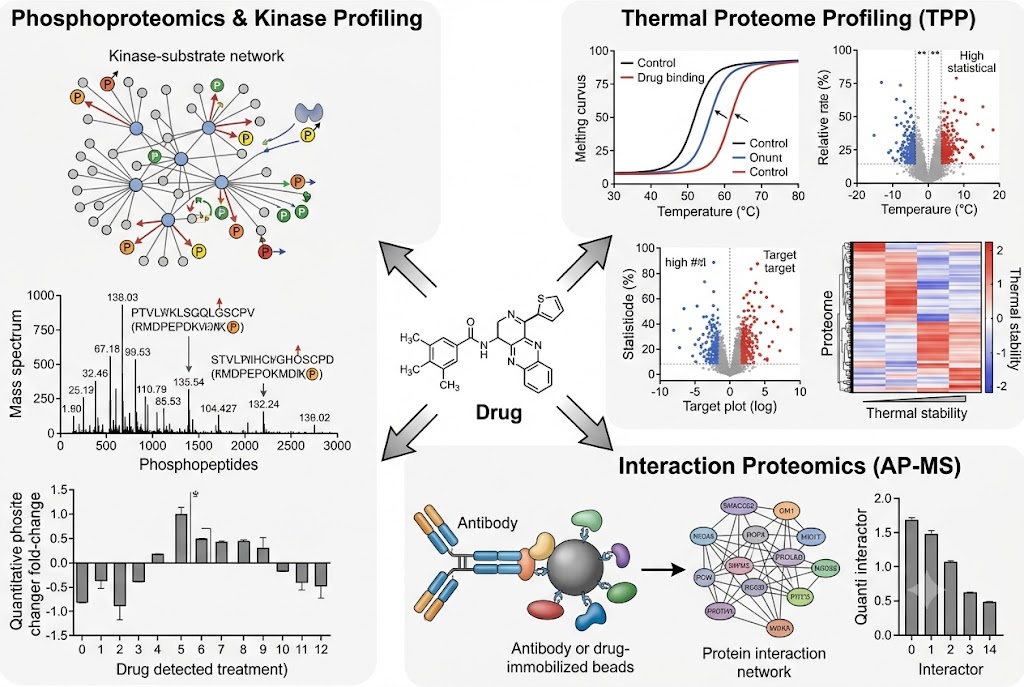
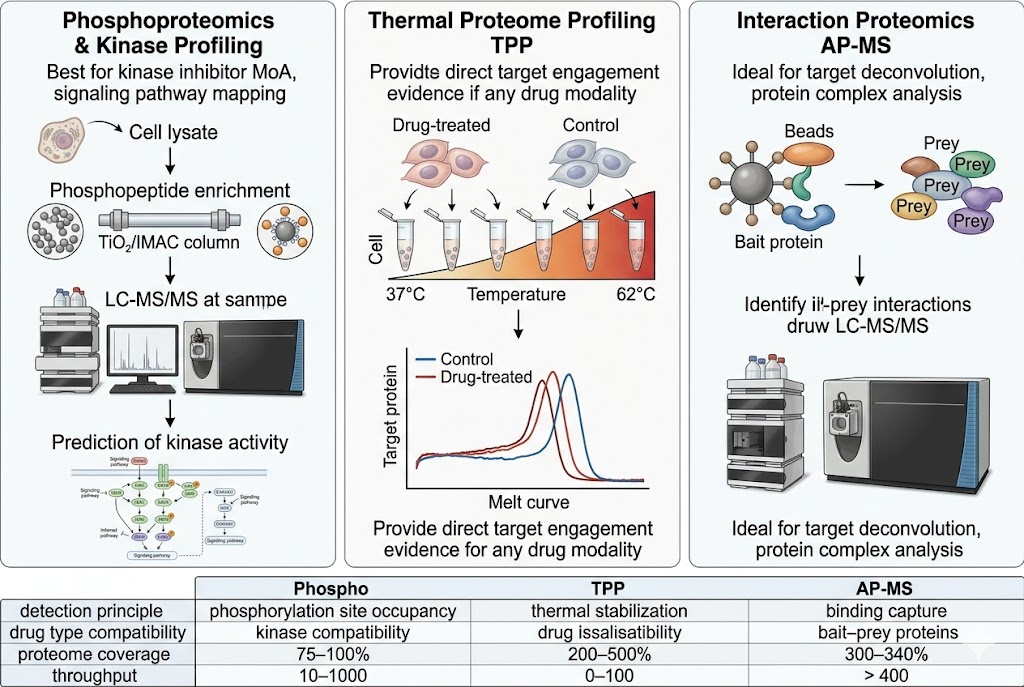
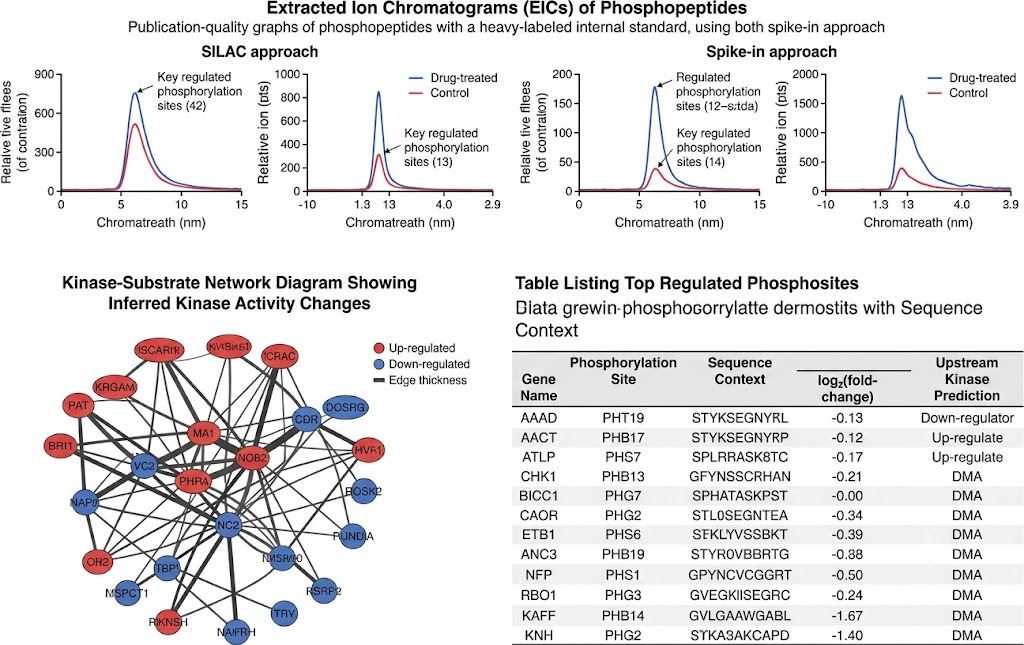
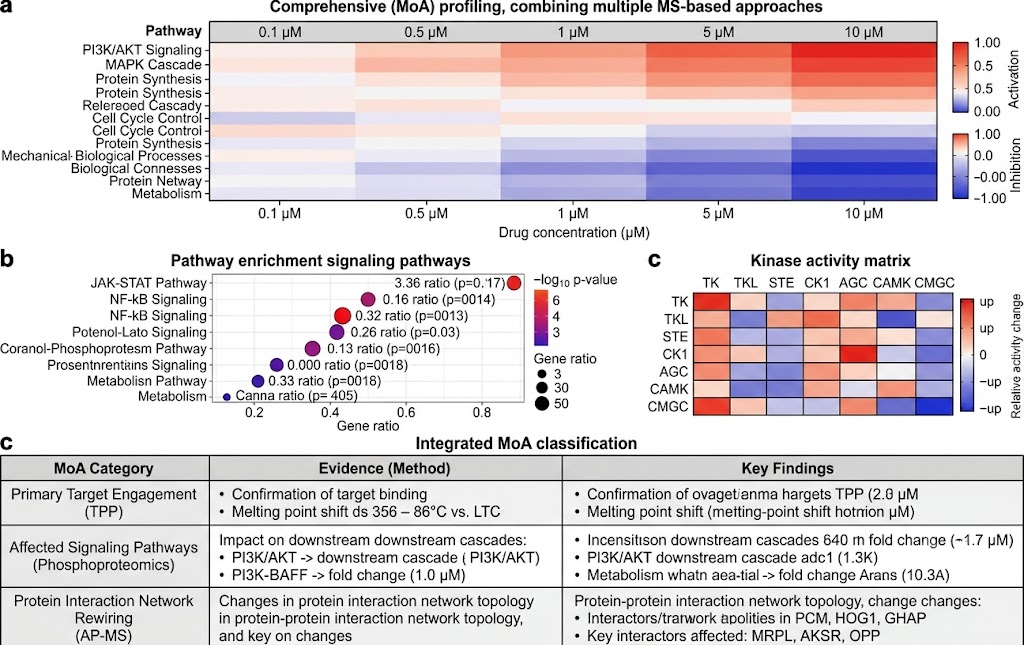
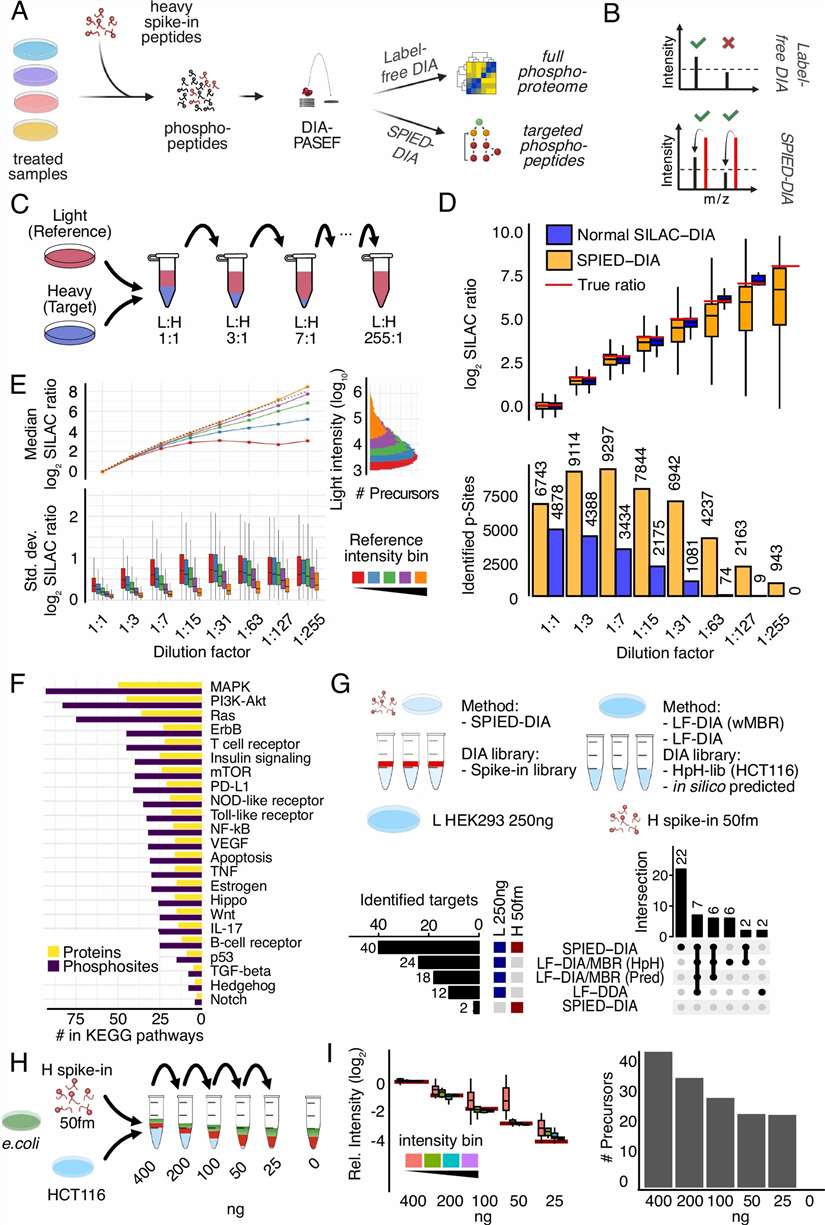
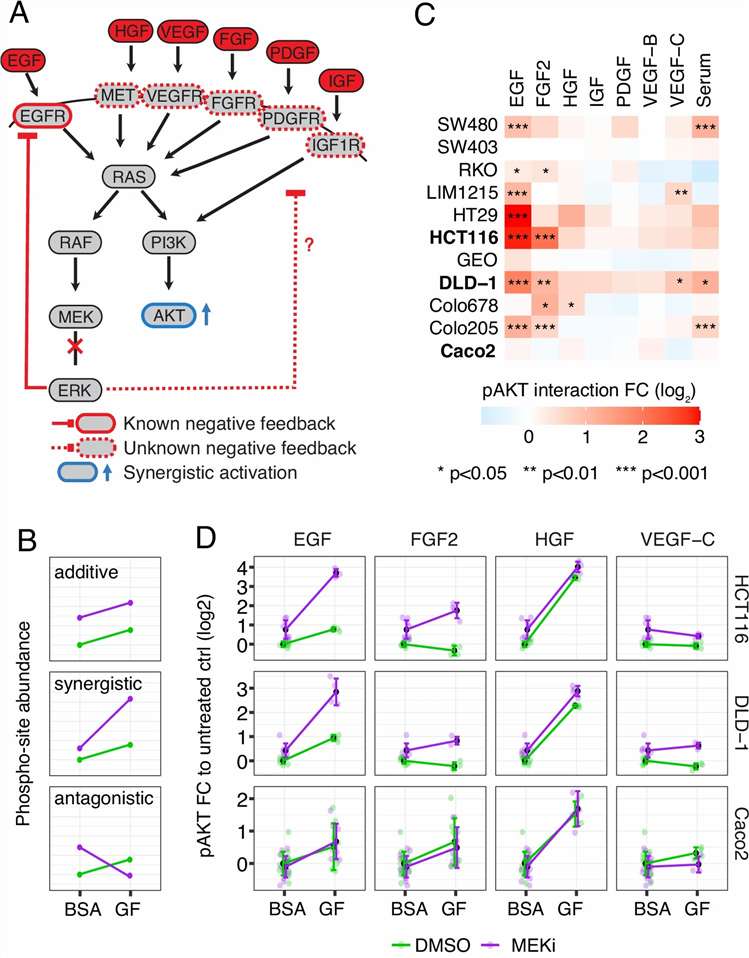
- Phosphoproteomics & Kinase Activity Profiling: Quantitative phosphoproteomics with TiO2/IMAC enrichment identifies thousands of regulated phosphorylation sites upon drug treatment. Kinase-substrate enrichment analysis and kinase activity inference algorithms translate phosphosite-level changes into pathway-level mechanistic understanding, revealing on-target pathway modulation, compensatory signalling, and resistance mechanisms. SILAC-based or label-free quantification with FDR-controlled statistics ensures high-confidence phosphosite assignment.
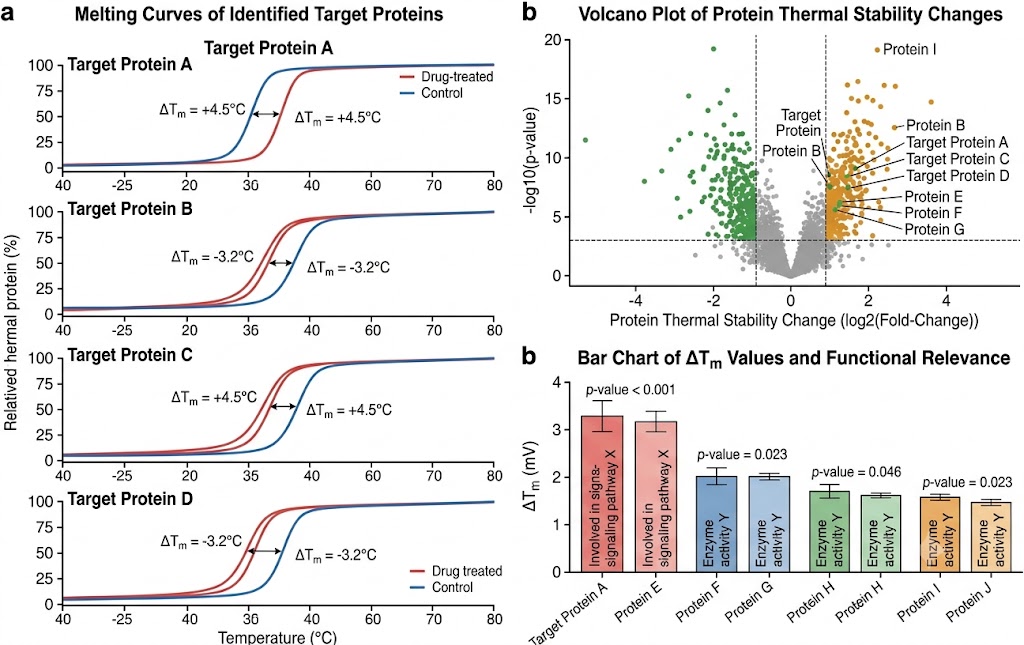
- Thermal Proteome Profiling (TPP): Multiplexed TMT-based TPP detects drug-induced changes in protein thermal stability across the proteome, providing direct biophysical evidence of target engagement in cellular context without requiring chemical modification of the drug. Ten-temperature gradient profiling with melt-curve fitting and ΔTm quantification for 6,000–8,000 proteins identifies both the intended target and additional proteins stabilised or destabilised by drug treatment.
- Interaction Proteomics (AP-MS): Affinity purification coupled with LC-MS/MS identifies drug-induced changes in protein-protein interaction networks. Antibody-based or epitope-tag pull-down of the target protein followed by quantitative proteomics distinguishes specific interactors from background, revealing drug-dependent complex assembly, disassembly, or recruitment that informs mechanism and potential toxicity.