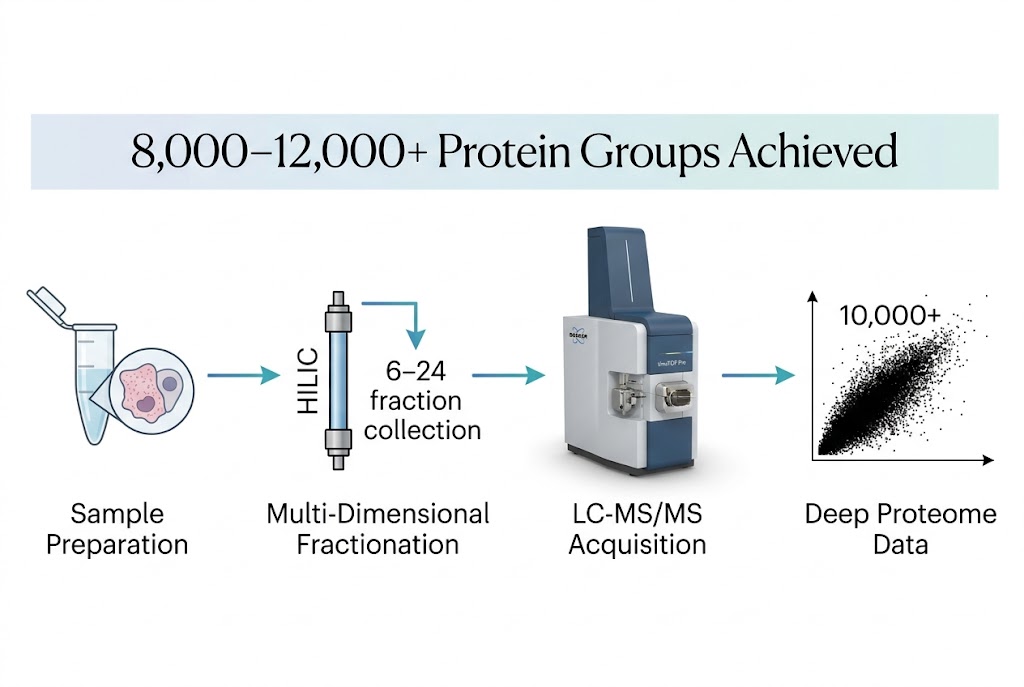
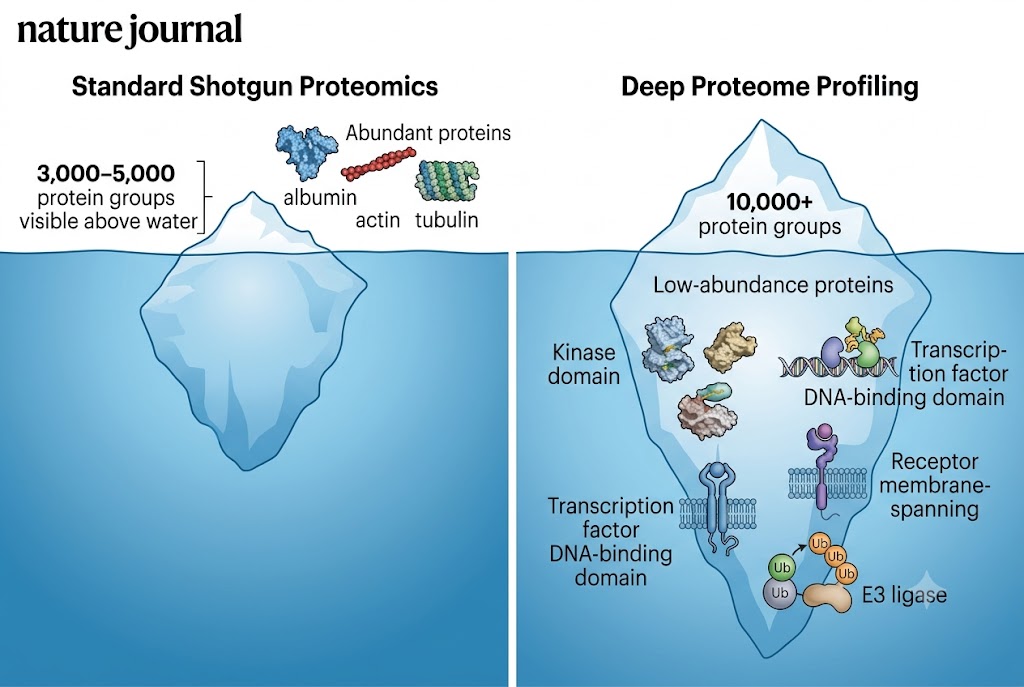
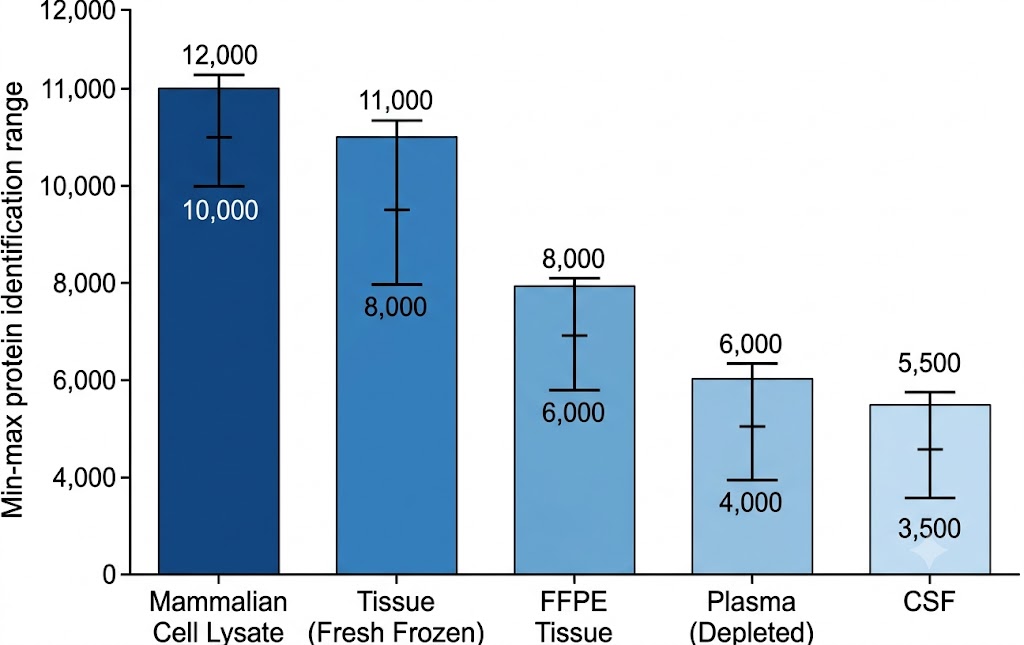
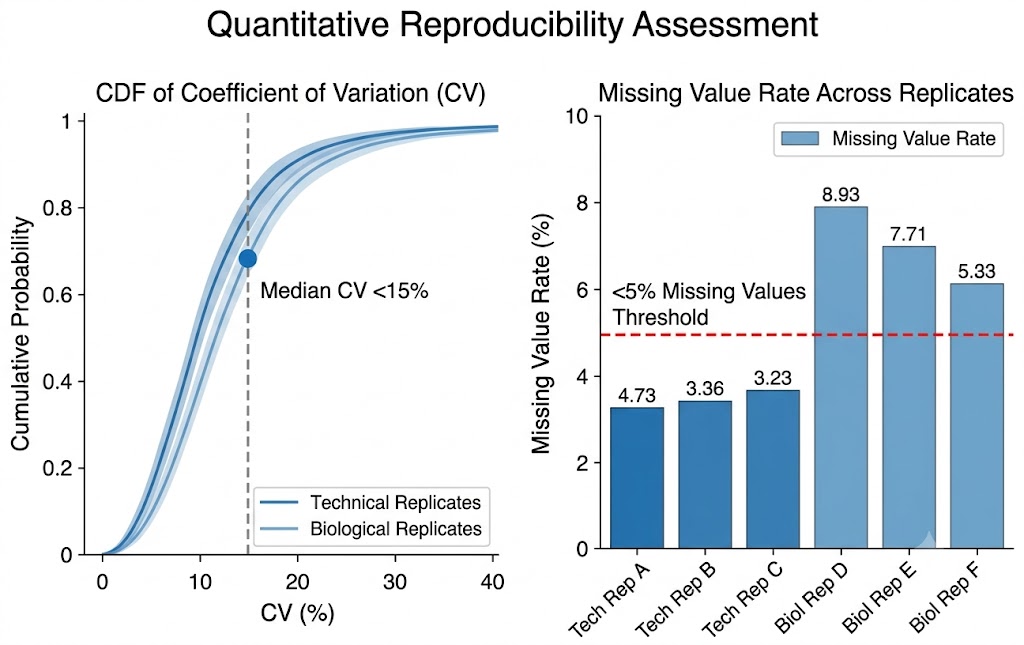
Biomarker Discovery Needs Depth — Low-Abundance Proteins Carry the Tissue-Specific Signal
The classical biomarker discovery paradigm holds that the most informative biomarkers are often the least abundant proteins: tissue-specific secreted proteins, shed receptor ectodomains, or signalling molecules that change concentration before abundant housekeeping proteins do. Yet standard plasma proteomics — even with high-abundance protein depletion — routinely identifies fewer than 500–800 proteins, dominated by albumin, immunoglobulins, and acute-phase reactants. Most tissue-derived candidate biomarkers are present in plasma at ng/mL concentrations or below, well below the detection limit of single-shot workflows.
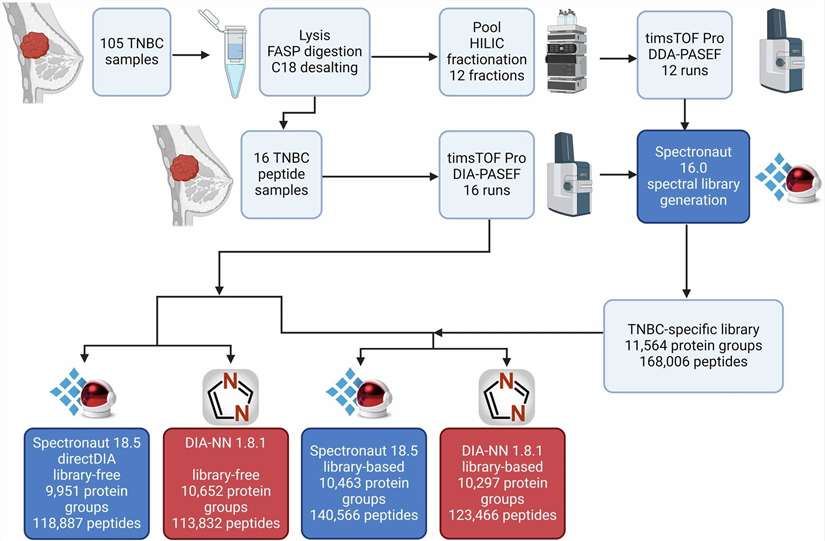
Deep Proteome Profiling addresses this through a combination of abundant protein depletion (where applicable), peptide-level fractionation, and DIA acquisition that together expand the detectable plasma proteome to 4,000–6,000 proteins. At this depth, tissue-specific proteins — brain-derived, cardiac, liver-secreted, tumour-associated — become detectable in the circulation. In published cohort studies using similar workflows, this depth expansion has directly translated to the identification of candidate biomarkers that were invisible to standard plasma proteomics, including proteins involved in neuronal signalling, extracellular matrix remodelling, and immune regulation — the very categories most likely to carry disease-specific information.
Drug Target Discovery Requires Access to the Regulatory Proteome
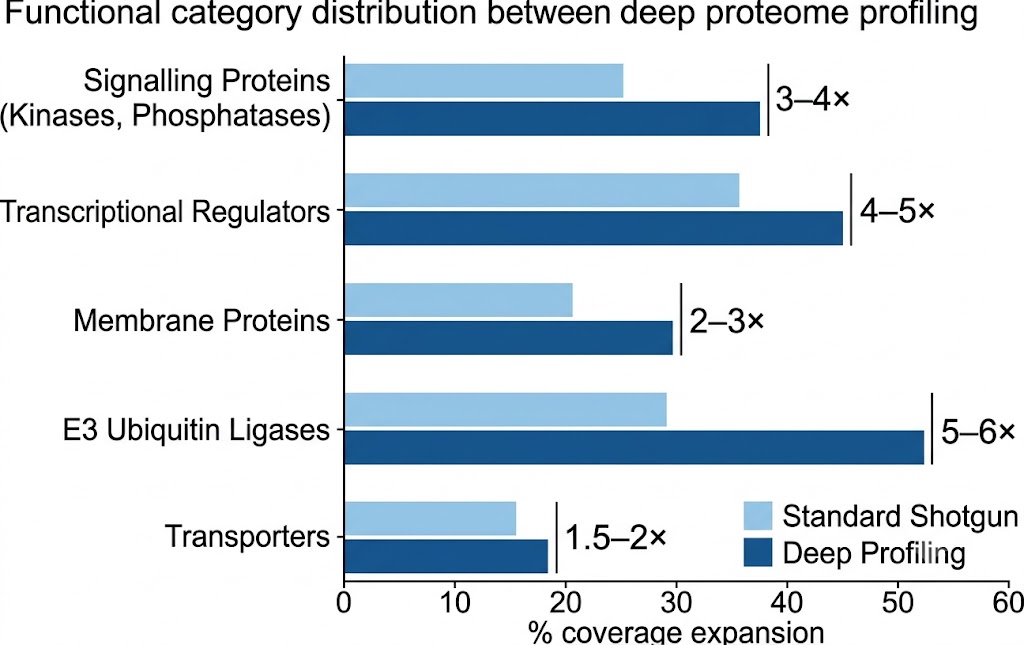
If your research question is mechanistic rather than descriptive — you want to know which signalling pathways are activated, which transcription factors drive the response, or which ubiquitin ligases regulate a substrate — then coverage of low-abundance proteins is not a luxury; it is a prerequisite. Kinase cascades, ubiquitin signalling networks, transcriptional regulation complexes, and protein-protein interaction modules are all executed by proteins expressed at copy numbers too low for single-shot shotgun proteomics to detect.
With deep profiling, we routinely identify 200–400 protein kinases and 50–150 transcription factors from a mammalian cell lysate — the difference between seeing the downstream readout (a phosphorylated protein) and seeing the upstream kinase that phosphorylated it. This depth enables target discovery scientists to map signalling networks from input to effector, not just from effector to phenotype.
Multi-Omics Integration Depends on Proteome Completeness
A common frustration in multi-omics studies is the asymmetry between transcriptome coverage (15,000–20,000 detected transcripts by RNA-seq) and proteome coverage (3,000–5,000 proteins by standard shotgun MS). This gap means that for every five genes that change at the RNA level, you have proteomics data for only one — making correlation analysis statistically unreliable and pathway-level interpretation heavily biased toward abundant proteins.
Deep Profiling closes this gap. At 10,000+ protein groups, the proteome-to-transcriptome mapping fraction rises to >50%, enabling meaningful correlation analysis across omics layers. Differentially expressed transcripts can be checked for corresponding protein-level changes; pathway enrichment can be computed on both omics layers with comparable coverage; and the integration analysis shifts from "what can we detect" to "what is biologically consistent."