Multi-Omics in HBV Infection
Although hepatitis B virus (HBV) poses a significant threat to global public health, the understanding of HBV-host interactions is limited. Based on ribosome profiling, SILAC, and RNA-sequencing analysis, the multi-omics landscape of the HBV-host interaction was generated, which will facilitate the development of anti-HBV treatment.
Hepatitis B Virus (HBV) and HBV infection
HBV infection is a common cause of liver disease globally, affecting more than 257 million people worldwide. Chronic HBV infection is also one of the major risk factors for hepatocellular carcinoma (HCC). As a major global public health threat, there is currently no complete treatment for HBV infection. HBV is a hepatotropic, non-cytopathic DNA virus. This virus can generate a covalently closed circular DNA (cccDNA) intermediate in the nucleus of infected cells. cccDNA plays an important role in the HBV life-cycle because it not only protects the virus from host pattern recognition factors but also acts as a transcriptional template for viral proteins. Targeting HBV cccDNA is considered to be an important strategy for the development of HBV therapeutic therapy. Currently, the treatment of chronic HBV infection mainly relies on type I interferon and nucleoside analogues (NAs) therapy. However, the former has strong toxicity and limited curative effect, while the latter cannot clear HBsAg (one of the viral antigens) and cannot achieve a functional cure.
Application of multi-omics in HBV infection
To better understand HBV infection, it is necessary to systematically elucidate the molecular details of host networks affected by HBV. Researchers used state-of-art-omics approaches to investigate HBV-host interaction, including ribosome profiling (riboseq), RNA-sequencing, and quantitative proteomic technology (Stable Isotope Labeling by/with Amino acids in Cell culture, SILAC).
Among them, riboseq can detect mRNA subsets of positive translation. This technique can map ribosome footprints on RNAs at nucleotide resolution and discover novel translational events, such as non-canonical open reading frames (ncorfs). Based on the combination of riboseq and SILAC, the effect of HBV on the translation of these ncorfs as well as their translation changes on HBV replication or viral gene expression were investigated. Conventional RNA-sequencing was applied to identify differentially expressed genes (DEGs) during HBV infection. A total of 35 canonical genes involved in transcription and translation (e.g., PPP1R15A, PGAM5, and SIRT6) and at least 15 non-canonical ORFs (e.g., ncPON2 and ncGRWD1) were identified during HBV infection. Particularly, SIRT6 has transcriptional cosuppressor activity related to gene silencing and showed transcriptional and translational downregulation during HBV infection. Moreover, MDL800 is a small molecular agonist of SIRT6 that showed the ability to inhibit HBV gene expression in both well-established cell and mouse models of HBV infection.
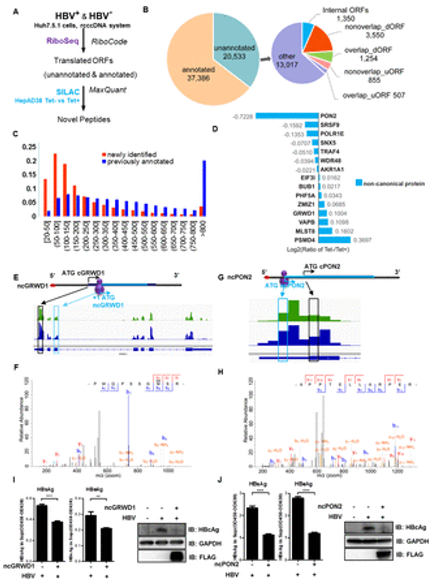 Fig1. Translation of non-canonical open reading frames (ncORFs) upon HBV replication. (Yuan, S., et al, 2021)
Fig1. Translation of non-canonical open reading frames (ncORFs) upon HBV replication. (Yuan, S., et al, 2021)
In order to develop potential diagnostics and prognostics as well as novel therapeutics to combat viral infection, more multi-omics approaches should be employed to provide a more comprehensive view of virus-host interactions. Creative Proteomics is a leading custom service provider in the field of viral multi-omics. We have a professional and experienced team capable of providing transcriptome, proteomics, metabolomics experiments, and multi-omics combined analysis services. Based on best-in-class technology and unmatched expertise, we are dedicated to providing the overall solution in the field of virus research from a multi-omics perspective. If you have any needs in this area, please feel free to contact us. We will serve you wholeheartedly.
Reference
- Yuan, S., et al. (2021). "Multiomics Interrogation into HBV (Hepatitis B Virus)-Host Interaction Reveals Novel Coding potential in Human Genome, and Identifies Canonical and Non-canonical Proteins as Host Restriction Factors against HBV." bioRxiv.
Related services
* For research use only.