Multi-Omics in Influenza A Virus Infection
Influenza A viruses (IAVs) are a major health threat to public health as they are capable of infecting different avian and mammalian animal hosts. Current treatment regimens are ineffective due to recurrent or newly emerging IAV variants occurring each year. Using multi-omics approaches, including genomics, transcriptomics, proteomics, and metabolomics, the novel modulators of IAV–host interactions can be identified, which will greatly facilitate the development of anti-influenza therapeutics. Here, let's get insight into the application of multi-omics in influenza A virus infection.
Influenza A virus (IAV)
Influenza is a contagious respiratory disease caused by influenza viruses (IVs) with single-stranded, negative-sense, and segmental RNA genomes. Influenza viruses evolve rapidly and can cause respiratory infections in both mammals and birds, including humans, pigs, chickens, and waterfowl. Based on the variations of the nucleoprotein (NP), the viruses can be divided into influenza A viruses (IAV), influenza B viruses (IBV), influenza C viruses (ICV), and influenza D viruses (IDV). Among them, IAVs belonging to the genus Alphainfluenzavirus, are of zoonotic importance. IAVs can be divided into a series of subtypes (including 16 HA- and 9 NA-subtypes) according to the antigenicity of the surface glycoproteins. All IAV subtypes were originally isolated from avian hosts except for bat-derived influenza-like viruses H18N11 and H17N10. The virus causes seasonal epidemics and sporadically pandemic outbreaks, posing an economic burden and health threat to humans.
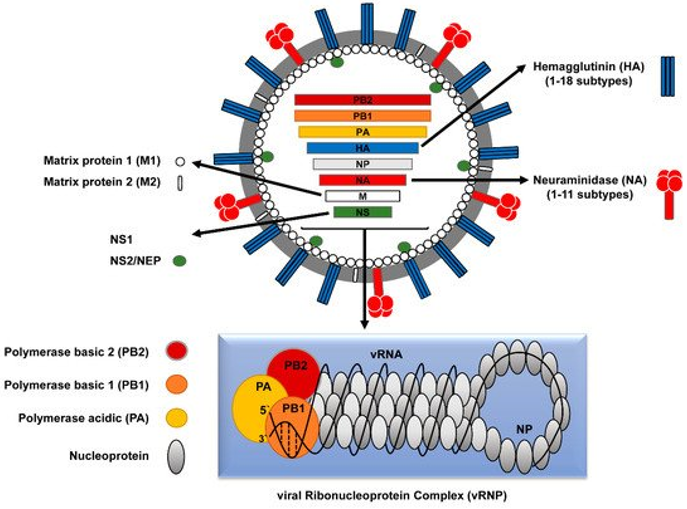 Fig1. Schematic structure of influenza A virus (IAV). (Mostafa, A., et al, 2018)
Fig1. Schematic structure of influenza A virus (IAV). (Mostafa, A., et al, 2018)
Multi-omics in influenza A virus infection—identification of potential cellular targets in IAV-host infection
In response to the rapid evolution of IAVs, it is necessary to develop novel antiviral agents to replace the conventional anti-influenza therapeutics. To improve the treatment options, more information about virus-host interactions is needed. Multi-omics approaches have been used to provide information about virus-host interactions at genetic, transcriptional, translational, and metabolic levels.
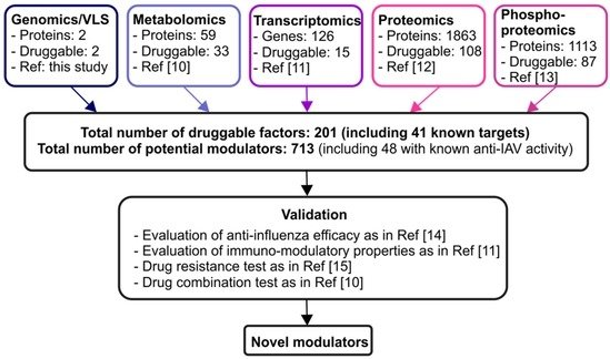 Fig2. Discovery pipeline for novel modulators of IAV-host interactions. (Söderholm, S., et al, 2016)
Fig2. Discovery pipeline for novel modulators of IAV-host interactions. (Söderholm, S., et al, 2016)
Application of transcriptomics in IAV infection
Based on the transcriptomics study, 126 genes were significantly altered in human peripheral blood mononuclear cells (PBMC)-derived macrophages infected with H3N2 and H1N1. There were several significant pathways associated with IAV infection, including cytokine signaling, cytokine-cytokine receptor interaction, interferon-α, -β, and -γ signaling, and cytosolic DNA-sensing pathway. Based on the Drug Bank and Drug Gene Interaction Database, 15 proteins that can be targeted with 53 compounds were identified.
Application of proteomics in IAV infection
Using the isobaric tags for relative and absolute quantitation (iTRAQ) technique, 2466 host proteins were quantified in human PBMC-derived macrophages infected with H3N2. Among them, 1321 proteins belong to the subcellular proteome and 544 in the secretome. Based on the Drug Bank and Drug Gene Interaction Database, there were 108 proteins that could be targeted with 346 compounds. Interestingly, TNF, CXCL10, CCL3, NAMPT, and CCL8 as druggable targets were both found in transcriptomics and proteomics study. In addition, 1113 phosphoproteins were altered upon infection and 87 of these proteins could be targeted by 382 compounds. Notably, 38 were also found among the druggable targets identified in the proteomics study.
Application of metabolomics in IAV infection
The metabolomics study using LC-MS/MS revealed that the metabolic profiles of PBMC-derived macrophages infected with H3N2 and H1N1. In addition to activated tryptophan catabolism, purine metabolism was modulated as a result of IAV infection, which were similar to transcriptomics experiments. Moreover, other alterations had been observed, such as glutathione, nitrogen, arginine, and proline metabolic pathways. Based on the Drug Bank and Drug Gene Interaction Database, there were 33 potential targets for 102 compounds.
If you have a question about our website or our services, please contact us.
References
- Mostafa, A., et al. (2018). "Zoonotic potential of influenza A viruses: a comprehensive overview." Viruses, 10(9), 497.
- Söderholm, S., et al. (2016). "Multi-omics studies towards novel modulators of influenza A virus-host interaction." Viruses, 8(10), 269.
Related services
* For research use only.