Proteomics in HIV Research
Due to its high throughput and sensitivity, proteomics has become a promising tool for HIV infection research, providing rich information on mechanisms underlying HIV pathogenesis, identifying biomarkers related to HIV infection, aiding in the diagnosis of HIV-related diseases, and so on. So far, there are multiple new proteins related to HIV infection discovered by using proteomic technology, such as CD44R5, ApoA1, CD14, and FGF-2. Creative Proteomics is an international contract research organization (CRO) providing viral proteomic, metabolomic and other analysis services to researchers in biochemistry, biotechnology, and biopharmaceutical fields. Here, we give an overview of several current proteomic technologies applied to investigate HIV infection.
It is very important to identify HIV-regulated host proteins
Despite much progress and efforts have been made in the last decade, human immunodeficiency virus (HIV)/acquired immune deficiency syndrome (AIDS) is still one of the major infectious diseases affecting global health. There is a need to identify new therapeutic targets and develop novel approaches. The viruses, including HIV, require multiple cellular elements to complete their life cycle. And these elements can be classified into four groups, including
- Membrane proteins for entry
- Cellular proteins for assembly, maturation, and transport
- Nuclear factor of transcription
- Viral regulatory and accessory proteins, which are associated with maintaining the viral life cycle
Since proteins are the ultimate effectors of most cellular functions, proteomic methods are considered to be one of the most productive ways to generate new information related to virus infection and have been widely used. Our understanding of the importance of proteomic technologies for more effective treatments and ultimately eradication of viral infections will benefit greatly from the above classification.
Proteomic technologies used in HIV studies
In recent years, a lot of proteomic technologies have been used to investigate HIV infection. These technologies include gel-based such as 2-dimensional electrophoresis (2DE), label proteomics such as isobaric tags for relative and absolute quantitation (iTRAQ) and amino acids in cell culture (SILAC), and label-free technologies. Furthermore, affinity purification-mass spectrometry (AP−MS) has been applied to identify proteins involved in HIV-host interaction, and newly-developed technologies for post-translational modifications (PTMs) have also been utilized in the study of HIV infection.
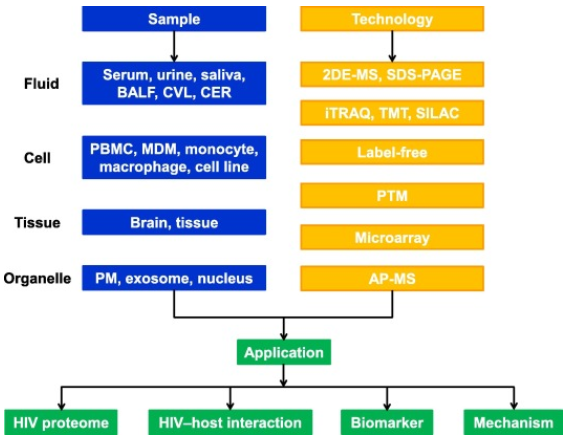 Fig1. Summary of current proteomic technologies and their applications in the study of HIV infection. (Zhang, L.,et al, 2017)
Fig1. Summary of current proteomic technologies and their applications in the study of HIV infection. (Zhang, L.,et al, 2017)
Here, we have shown the advantages and disadvantages of several current proteomic technologies in HIV studies. Importantly, we provide a series of proteomic techniques to help customers quickly and comprehensively investigate viral proteomics. Our services include three major parts, including viral protein structure detection, global proteome profiling, mass spectrometry-based viral proteomics. We are committed to providing you with the best strategy according to the specific project needs of our global clients.
| Technique | Advantage | Disadvantage |
| Gel-based methods, such as 2DE and SDS-PAGE | Samples processed ex vivo; able to quantify isoforms/PTMs | Labor-intensive and lower throughput; limited to pair-wise comparisons |
| SILAC | Stably integrate into all proteins | Limited to cultured cells or animal tissues |
| iTRAQ | Suitable for all kinds of samples and able to label up to eight samples in a single experiment | Co-selection of peptides by mass spectrometer leading to reporter ion intensity suppression |
| TMT | Suitable for all kinds of samples and able to label up to ten samples in a single experiment | Co-selection of peptides by the mass spectrometer leading to reporter ion intensity suppression |
| MRM/SRM | Absolute targeted quantitation; high throughput | Difficult to identify consistent peptides to track for quantitation |
| Label-free quantitation | No need for expensive isotope labels; able to analyze individual samples; broad adaptability | Require good replication for mass spectrometry and sophisticated software for data interpretation |
| AP-MS | Direct study of protein-protein interaction | Unable to find different proteins unless combined with other technologies |
| Microarray | Avoid problems with protein purification and stability; High throughout; samples processed ex vivo | Require many kinds of antibodies; expensive |
| SWATH-MS | No need for isotopic labeling; suitable for the study of various samples; higher coverage and accuracy; the data can be archived and analyzed retrospectively to achieve maximal protein identification | Expensive; Require sophisticated software for data interpretation |
If you are unable to find the specific service you are looking for, please feel free to contact us.
References
- Zhang, L.,et al. (2017). "Recent 5-year findings and technological advances in the proteomic study of HIV-associated disorders." Genomics, proteomics & bioinformatics, 15(2), 110-120.
- Donnelly, M. R., & Ciborowski, P. (2016). "Proteomics, biomarkers, and HIV-1: A current perspective." PROTEOMICS-Clinical Applications, 10(2), 110-125.
Related services
* For research use only.