Viral SWATH-MS Proteomics Service
Creative Proteomics is an international contract research organization (CRO) providing viral proteomic, metabolomic and other analysis services to researchers in biochemistry, biotechnology, and biopharmaceutical fields. We have launched a sequential window acquisition of all theoretical mass spectra (SWATH-MS) proteomic platform. SWATH-MS is a specific variant of data-independent acquisition (DIA) methods. As a label-free application of mass spectrometry (MS), this technology combines deep proteome coverage capabilities with quantitative consistency and accuracy. Therefore, SWATH-MS is suitable for long-term projects or projects with large sample sets which require accurate and high reproducible quantitation.
Overview of sequential window acquisition of all theoretical mass spectra (SWATH-MS)
The SWATH-MS method, first described by Gillet et al, involves a target data extraction strategy for DIA spectral library mining SWAS-MS data. SWATH-MS is now emerging as a powerful technology for the discovery of proteins within complex biological mixtures. Unlike traditional MS which is limited in the discovery-based proteomics within the entire range of the spectrum, this method divides the acquired spectrum into successive segments and collects all information within each segment. Therefore, it theoretically allows the identification and quantification of all peptides in the specific analyte.
In a SWATH-MS experiment, all ionized peptides of a given sample are fragmented across SWATH windows in a systematic and unbiased manner and then analyzed in MS/MS, enabling sensitive and accurate quantitation of all ionized peptides, even for low abundant ones. SWATH requires the preconstruction of a spectral library that contains MS coordinates for each target peptide using traditional data-dependent acquisition. Based on the information, proteins presented in the library can be identified and quantified by SWATH-MS. In total, the experimental groups are performed by a data-independent mode of MS, followed by analyzed and identified against the preconstructed library after data acquisition. Therefore, the data can be archived and analyzed retrospectively to achieve maximal protein identification as the spectral libraries are further improved.
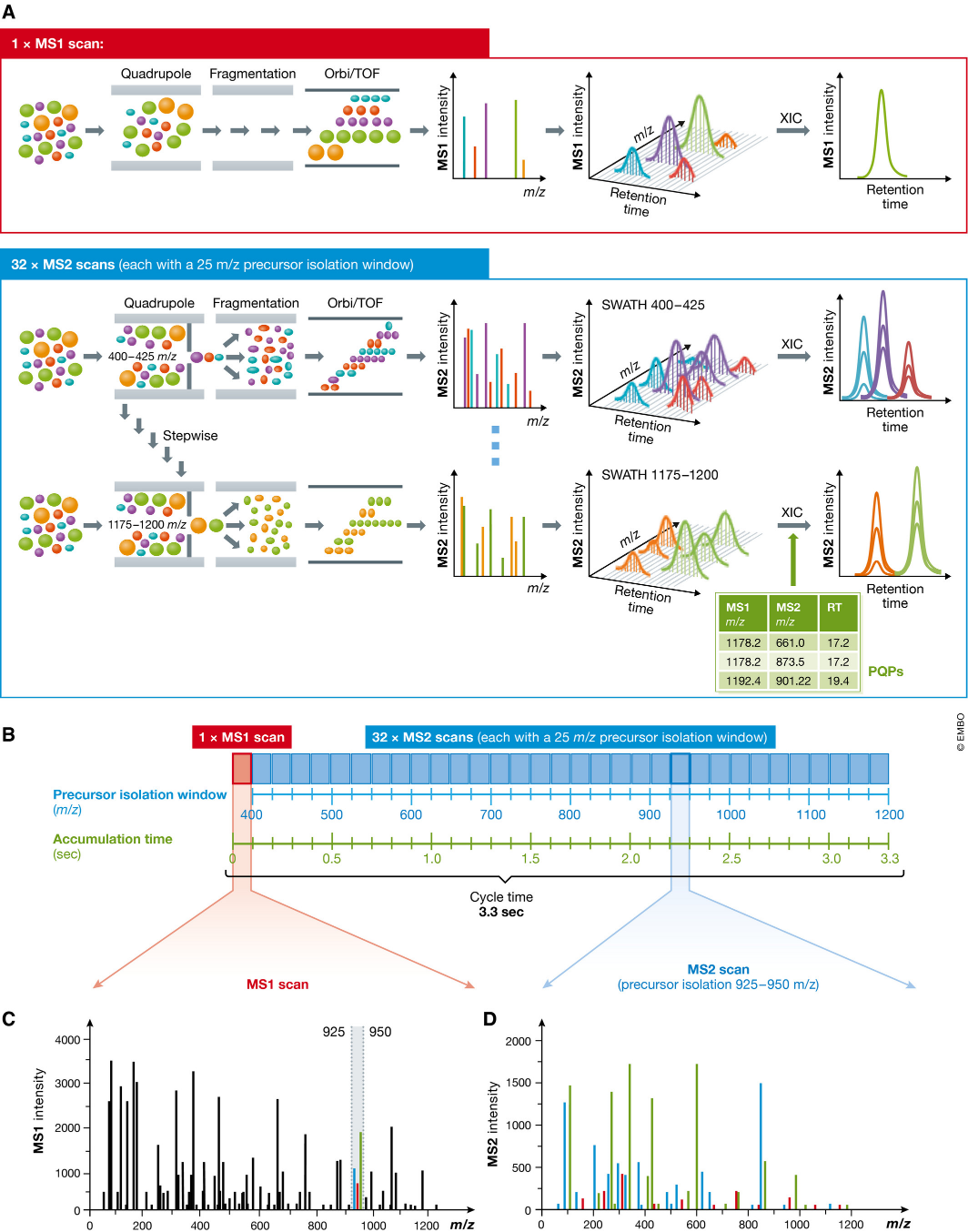 Fig1. Principle of sequentially windowed data-independent acquisition in SWATH-MS. (Ludwig, C., et al, 2018)
Fig1. Principle of sequentially windowed data-independent acquisition in SWATH-MS. (Ludwig, C., et al, 2018)
Benefits of label-free quantitative proteomics
- Higher coverage and accuracy.
SWATH-MS combines the high throughput of the shot-gun method with the repeatability of the selected reaction monitoring (SRM) method.
- No need for isotopic labeling.
- Less sample processing and high operating efficiency of software and database.
- Strict quality control. The data can be archived and analyzed retrospectively to achieve maximal protein identification.
- Suitable for the study of various samples, such as subcellular structure, bacteria, and viruses.
Service offering
We focus on the continued development of data-independent acquisition mass spectrometry (DIA-MS), especially the SWATH-MS approach. Based on experienced scientists and well-established platforms, we strive to stay on the leading edge of virus- related protein identification and quantification. Our SWATH-MS proteomic platform enables large-scale sample analysis while maintaining a high protein quantity and accuracy. Customers only need to send their samples to us, and we will take care of all the follow-up matters of the project, including protein extraction, protease digestion, peptide separation, MS analysis, raw data and bioinformatics analysis.
How to order?

Creative Proteomics specializes in quantitative and qualitative proteomics and metabolomics applications based on state-of-the-art MS platforms, coupled with high-performance liquid chromatography technology. Combined with subcellular fractionation and SWATH-based MS, we can help global customers to map the global intracellular changes induced by virus infection through quantifying protein abundance within the subcellular compartments of uninfected and virus-infected cells.
If you are interested in our services, please don't hesitate to contact us. We are glad to cooperate with you and witness your success!
Reference
- Ludwig, C., et al. (2018). "Data-independent acquisition-based SWATH-MS for quantitative proteomics: a tutorial." Molecular systems biology, 14(8), e8126.
* For research use only.