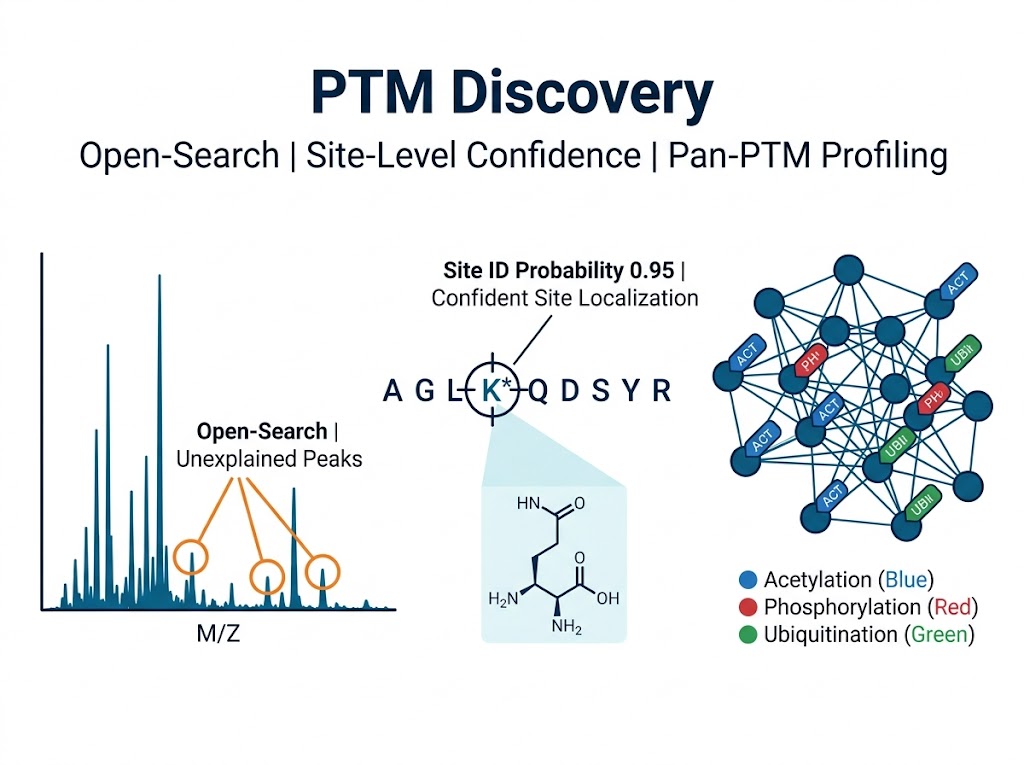
Why PTM Discovery — From Hypothesis to Modification Landscape
The most valuable PTM experiments are the ones that find modifications you did not design the experiment to find. A targeted assay confirms what you suspect. Discovery, by contrast, asks an open question: what modifications exist in this sample, at which sites, and with what relative abundance?
We deploy three complementary discovery modes. Open discovery — no predefined modification list; the search engine considers a wide mass range, detecting unanticipated modifications including oxidation products, drug metabolite adducts, and novel acylations. Selected discovery — targeted enrichment for a specific modification class (phosphorylation, acetylation, ubiquitination) followed by comprehensive site identification across the proteome. Pan-PTM discovery — parallel enrichment for multiple modification families from the same sample, enabling direct comparison of modification crosstalk at shared lysine residues. Each mode produces site-level quantification with the confidence metrics needed to prioritize follow-up validation.
Unknown & Low-Abundance PTM Discovery — Open-Search Strategies for Novel Modifications
Standard database search pipelines define a finite set of variable modifications — typically 3–6. Any peptide carrying a modification outside this set is either not identified or incorrectly assigned. Our open-search workflow removes this constraint.
How it works. Using MaxQuant with dependent peptides or pFind Studio's open-search mode, we search MS/MS spectra against the proteome database with an unrestricted precursor mass window (typically ±200–500 Da). Unmatched spectra are re-searched against a broader modification database including over 200 documented PTMs. A second pass uses a sequence tag–based approach (Open-pFind) that builds candidate peptide sequences directly from fragmentation spectra and identifies any mass shift as a potential modification.
What it detects. This approach is how lactylation, 2-hydroxyisobutyrylation, and monoaminylation were identified — each initially detected as an unexplained mass shift in an open-search experiment. In our hands, open-search workflows identify 15–40 distinct modification types per project, with 3–8 being modifications not included in the initial search parameter set.
Our dedicated Open-Search PTM Discovery service provides full details on search parameters, instrument requirements, and expected output.
PTM Site Identification & Localization — Confidence, Evidence, and False Discovery Control
A modification site is only as useful as the confidence that it is correctly assigned. Our pipeline addresses three factors that determine site-level confidence:
Localization probability. Using MaxQuant's position probability algorithm, each potential site within a peptide is assigned a score (0–1). Sites with probability ≥0.75 are reported as localized; ≥0.95 as high-confidence. The probability difference between the top-ranked and second-ranked position must exceed 0.1.
Fragment ion coverage. For every reported modified peptide, we verify that site-determining fragment ions are present in the MS/MS spectrum. Spectra lacking site-determining ions are flagged and excluded from high-confidence reporting.
FDR control. FDR is controlled at three levels: PSM level (<1%), peptide level (<1% via target-decoy), and site level (class I: localization probability ≥0.75). This three-tier framework ensures reported modification sites are not artifacts of incorrect peptide assignment.
For projects requiring deep validation of specific targets, our PTM Site Identification service provides extended gradient and targeted MS/MS acquisition for low-abundance modified peptides.
Global & Pan-PTM Profiling — Selected Modification versus Multi-Modality Discovery
Researchers studying PTM biology face a strategic choice: invest depth in a single modification type, or survey breadth across multiple types. Our platform supports both approaches.
Global selected PTM profiling focuses on maximum depth for a single modification class. Using modification-specific enrichment (antibody-based or chemical) followed by deep LC-MS/MS, this approach identifies 5,000–15,000 modification sites per project with the deepest possible coverage for comprehensive site discovery and differential analysis.
Pan-PTM multi-modality profiling performs parallel enrichment for multiple modification classes from aliquots of the same digested sample. A typical project covers acetylation, phosphorylation, ubiquitination, and one or two additional modifications. The output enables direct comparison of modification occupancy at individual residues, revealing crosstalk patterns that single-modification studies miss.
Our Global PTM Profiling and Pan PTM Proteomics services provide detailed workflows and project examples.
PTM proteoform mapping. Beyond site identification, we offer proteoform-level analysis using middle-down proteomics and intact protein analysis to determine which combinations of modifications co-occur on the same protein molecule. This is particularly relevant for histone modification combinations. See our PTM Proteoform Mapping service.
Emerging PTM Discovery — Finding the Next Layer of Regulatory Modifications
The PTM landscape has expanded dramatically in the last decade. Modifications that were unknown or considered rare in 2015 — lactylation, crotonylation, 2-hydroxyisobutyrylation, β-hydroxybutyrylation, monoaminylation — are now recognized as physiologically significant regulators. Our emerging PTM discovery service detects modification types that fall outside standard search parameters.
Three independent detection approaches: (1) Open-search MS identifies unexplained mass shifts and assigns them to candidate modification types. (2) Chemical derivatization provides orthogonal confirmation via selective labeling. (3) Biological validation — correlation with metabolic precursors (lactate, β-hydroxybutyrate, serotonin) — confirms the modification responds to physiological stimuli.
Recent examples. We have identified lactylation in cancer cell lines under glycolytic conditions, detected crotonylation in HDAC inhibitor-treated cells with characteristic H3K18cr enrichment, and mapped 2-hydroxyisobutyrylation on metabolic enzymes in pancreatic cancer tissue — each initially discovered through open-search and subsequently validated.
For first-pass assessment, our PTM Qualitative Analysis service provides a rapid survey option.
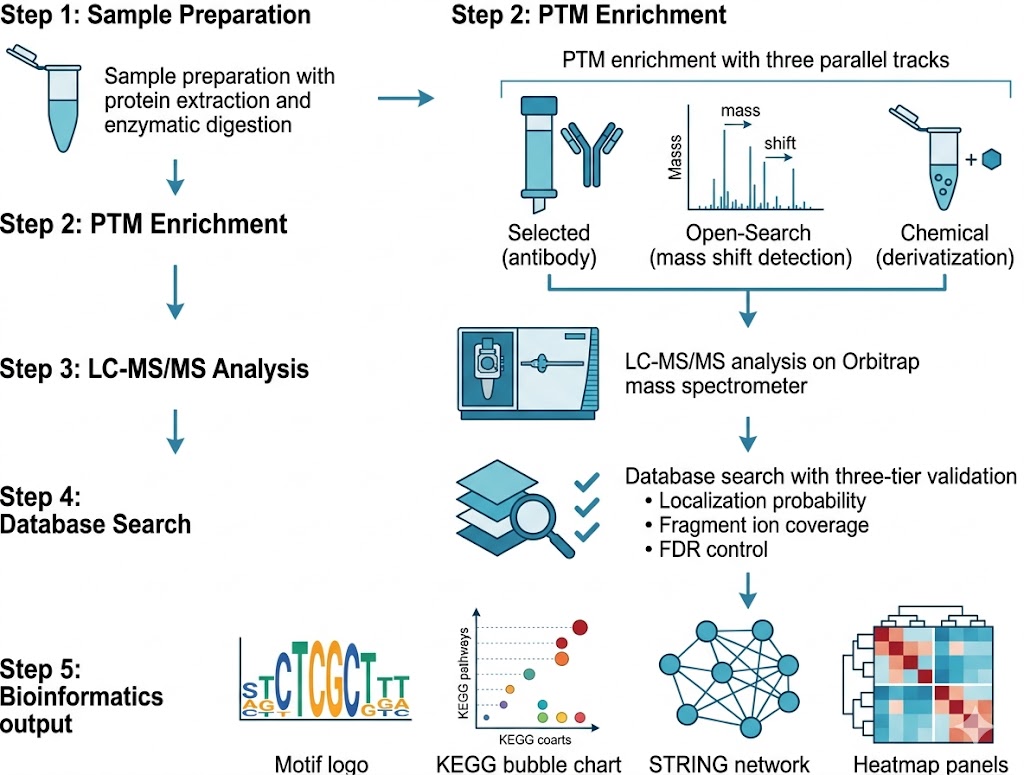
Publication-Ready Bioinformatics — Motif, Pathway, Network, and Crosstalk Analysis
Raw PTM site lists have limited value for biological interpretation. Our bioinformatics pipeline transforms identification data into publication-ready formats across five analysis tiers:
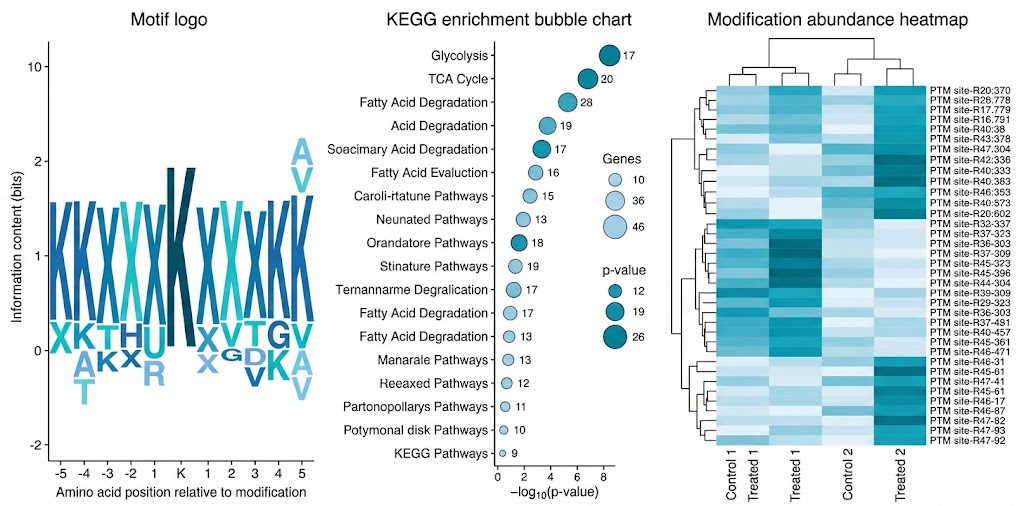
Sequence motif analysis. Using pLogo or IceLogo, we analyze the ±10 amino acid window around each modification site to define sequence preference. This reveals modification-specific consensus motifs that provide insight into the responsible writer enzyme.
GO and pathway enrichment. GO enrichment identifies biological processes and molecular functions overrepresented among modified proteins. KEGG pathway mapping highlights signaling and metabolic pathways where the modification is concentrated, answering: "what biology does this modification regulate?"
Interaction network analysis. Using STRING, we map modified proteins onto interaction networks. PTM-enriched modules identify functional complexes that are preferentially modified, revealing pathway-level regulatory logic.
PTM crosstalk analysis. For multi-modality projects, we identify residues carrying more than one modification type, quantify enrichment or depletion of co-occurring modifications, and generate correlation matrices — particularly valuable for understanding acetylation, phosphorylation, and ubiquitination competition at shared residues.
Publication-ready figures. All standard outputs delivered as vector PDF and 300 dpi PNG: motif logo plots, GO/KEGG enrichment bubble charts, modification abundance heatmaps, volcano plots, and STRING networks with modified nodes highlighted.
Frequently Asked Questions About PTM Discovery
Q: What is the difference between global PTM profiling and pan-PTM proteomics?
A: Global PTM profiling focuses on maximum depth for a single modification type. Pan-PTM proteomics simultaneously profiles multiple modification types from the same sample for direct crosstalk comparison. Choose based on whether your question is depth (global) or breadth (pan-PTM).
Q: How confident can I be in the reported modification sites?
A: We report localization probability, fragment ion coverage, and FDR at three levels (PSM, peptide, site). Only sites with probability ≥0.75 confirmed by site-determining fragment ions are included in the high-confidence output.
Q: Can you discover modifications that have not been previously reported?
A: Yes. Our open-search pipeline detects unexplained mass shifts in MS/MS spectra. This is the same approach that led to the discovery of lactylation, crotonylation, and other recently described PTMs.
Q: How much sample is needed for a PTM discovery project?
A: For global profiling of a single modification: 1–5 mg total protein. For pan-PTM profiling (3–5 modifications): 5–15 mg. For open-search discovery without enrichment: 100–500 μg.
Q: Can I combine PTM discovery with quantitative comparison across conditions?
A: Yes. Label-free quantification is included in all discovery projects. For higher precision, TMTpro-based multiplexing (up to 18-plex) or SILAC can be integrated.
Q: What is the typical timeline for a PTM discovery project?
A: Global profiling of a single modification: 4–6 weeks. Pan-PTM profiling: 6–8 weeks. Open-search discovery: 3–4 weeks.
References
- Olsen JV, Blagoev B, Gnad F, et al. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 2006;127(3):635-648. doi:10.1016/j.cell.2006.09.026
- Choudhary C, Kumar C, Gnad F, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325(5942):834-840. doi:10.1126/science.1175371
- Zhang H, Tang K, Luo J, et al. Global landscape of 2-hydroxyisobutyrylation in human pancreatic cancer. Frontiers in Oncology. 2022;12:1001807. doi:10.3389/fonc.2022.1001807
*This service is provided for research use only (RUO). It is not intended for clinical diagnostic or therapeutic applications.