What Is Citrullination
Citrullination, catalyzed by peptidylarginine deiminases (PADs), is an irreversible process in which arginine residues within a protein peptide chain are converted into citrulline residues. This post-translational modification plays a crucial role in various biological processes. It impacts hydrogen bond formation, protein folding, hydrophobicity, and protein-protein interactions, ultimately leading to protein denaturation. Common substrates for citrullination by PADs include keratins, myosin, vimentin, actin, histones, collagen, and myelin basic protein. Notably, free arginine residues are not susceptible to citrullination by PADs.
Citrullination of histone arginines is a significant epigenetic mark that plays a pivotal role in chromatin remodeling and the extracellular trapping process of immune cells. Citrullination of arginines at positions 2, 8, and 17 of histone H3 is primarily catalyzed by peptidylarginine deiminase 4 (PAD4). Citrullination of histones, particularly histone H3 citrullination, has been revealed as a comprehensive manifestation of diverse inflammatory signals triggering neutrophil responses to infections.
It has been reported that citrullinated histone H3 may serve as a potential serum biomarker for early diagnosis of septic shock. Furthermore, alterations in histone H3 citrullination appear to be associated with immunological diseases such as multiple sclerosis and rheumatoid arthritis. Modulation of total histone H3 citrullination can be achieved by inhibiting or activating PAD enzymes. Therefore, quantitative assessment of total histone H3 citrullination offers valuable insights into understanding epigenetic regulation of gene activation and silencing, as well as the development of PAD-targeted therapeutics.
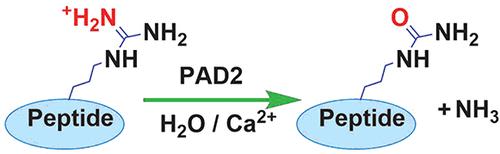 Citrullination reaction catalyzed by PAD2 (Erdem Cicek et al. Biochemistry 2022)
Citrullination reaction catalyzed by PAD2 (Erdem Cicek et al. Biochemistry 2022)
Ms-Based Analysis Of Citrullinated Proteins
To gain a comprehensive understanding of the role of citrullination, it is crucial to identify both citrullinated proteins and the specific citrullination sites. Mass spectrometry-based proteomics is the method of choice for achieving this goal.
Mass spectrometry stands out as the essential tool uniquely capable of precisely determining citrullination sites within proteins. However, the challenge lies in the subtle mass alteration of only 0.98 Da resulting from citrullination of an arginine residue. This challenge arises from the difficulty of distinguishing this mass shift from other potential factors, such as 13C isotopes or common deamidation events in asparagine (Asn) and glutamine (Gln) residues. Additionally, citrullination eliminates a trypsin cleavage site, leading to longer peptides that are challenging to detect via tandem mass spectrometry. These complexities become more pronounced when analyzing low-abundance proteins.
To address these challenges, researchers have adopted innovative approaches. For example, some have used collision-induced dissociation (CID) in combination with high-energy collision dissociation (HCD). CID induces the neutral loss of isocyanic acid from citrullinated peptides, while HCD facilitates the detection of lower mass-to-charge ratio (m/z) ions. This approach has enabled the identification of citrullination sites within various protein substrates of peptidyl arginine deiminases (PADs), without specific examples. Recent efforts have aimed at comprehensive proteomic investigations across various human tissues using the same strategy, revealing numerous citrullination sites across different proteins. However, the majority of identified proteins were of high abundance, highlighting the need for improved methodologies capable of selectively enriching low-abundance citrullinated proteins before their detection.
Enrichment Strategies for Citrullinated Proteins
Citrulline Reactive Beads (CRB): Tutturen et al. introduced a method utilizing CRBs. These beads are designed to specifically capture citrullinated peptides. The process involves immobilizing citrullinated peptides on the CRB, washing away non-citrullinated peptides, and subsequently eluting the bound citrullinated peptides. The enrichment method has shown promise in enriching citrullinated peptides, although background signals from bead-related substances can be a limitation.
Biotin-Based Enrichment (BPG): Another enrichment strategy developed by Tutturen and colleagues involves the use of biotin-PEG-GBA (BPG) tags [103]. Peptides are first tagged with BPG, and unreacted tags are removed. Streptavidin beads are then used to capture the tagged peptides, and subsequent elution with excess biotin allows for the recovery of the enriched citrullinated peptides. This approach offers cleaner enrichment, without the background interference observed with CRBs, but it requires multiple steps for tag synthesis.
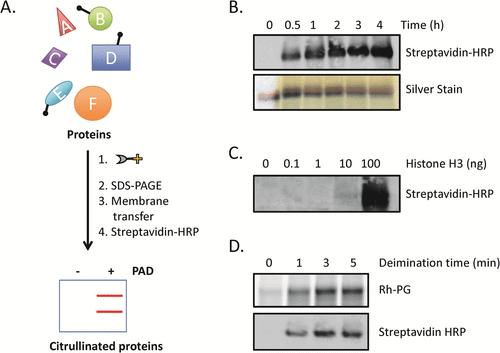 Using biotin-PG to visualize protein citrullination (Daniel M. Lewallen et al. ACS Chemical Biology 2015)
Using biotin-PG to visualize protein citrullination (Daniel M. Lewallen et al. ACS Chemical Biology 2015)
These enrichment strategies have proven effective in improving the identification of citrullinated peptides within complex mixtures. They provide a means to selectively enrich for citrullinated peptides, thereby increasing their concentration relative to non-modified peptides before analysis. These strategies represent significant advancements in the field of citrullination analysis, enabling the identification of citrullinated proteins, including those of lower abundance, and enhancing our understanding of the role of citrullination in various biological processes and diseases.
Citrullinated Proteins Analysis Service at Creative Proteomics
Creative Proteomics offers comprehensive mass spectrometry-based citrulline quantitation and modification site analysis services, covering all aspects from sample preparation to data analysis. Whether you require single-site analysis or large-scale investigations, our services are tailored to meet your specific needs.
Protein Sample Preparation: Prior to analysis, biological specimens, including tissues and bodily fluids, undergo meticulous processing to effectively extract and solubilize proteins. Ensuring proper sample preparation procedures is paramount to preserving the structural integrity of citrullinated proteins.
Mass Spectrometry Analysis: State-of-the-art mass spectrometry instrumentation is employed to facilitate the identification and characterization of citrullination sites. Leveraging advanced technologies such as electron transfer dissociation (ETD) and multiple reaction monitoring (MRM) ensures the achievement of precise and highly sensitive results, thereby enhancing the accuracy of citrullination site determination in a manner consistent with scientific and academic writing conventions.
Data Analysis: Our service capitalizes on cutting-edge bioinformatics tools and specialized software to methodically process and meticulously interpret mass spectrometry data. Our data analysis extends to functional assessments, such as pathway analysis, gene ontology (GO) analysis, and protein-protein interaction network analysis, providing comprehensive insights into the biological significance of citrullination events.
Our Detection Methods for Citrullinated Protein Analysis
In the pursuit of citrullination analysis, we select the best method based on your needs to address its inherent challenges.
| Methodology |
Description |
| Combination of CID and HCD |
Collision-induced dissociation (CID) combined with high-energy collision dissociation (HCD) has been employed for the identification of citrullinated peptides. CID triggers the neutral loss of isocyanic acid, while HCD enables the detection of lower m/z ions. This synergistic strategy substantially improves the precision of citrullination site identification. |
| SILAC Quantification |
For quantitative analysis, we have utilized Stable Isotope Labeling by/with Amino Acids in Cell Culture (SILAC). This method entails the cultivation of cells in media with distinct isotopic compositions, thereby facilitating precise measurement of the relative abundance of citrullinated proteins. |
| DIA Quantitative Proteomics |
To achieve large-scale quantitation of citrullination, we employed data-independent acquisition (DIA), specifically the sequence window acquisition of all theoretical fragment ion spectra. This DIA proteomics methodology offers extensive coverage of the citrullinome and unveils variations in citrullination levels across diverse experimental conditions or in the context of disease states. |
Advantages of Our Service
Precision in Site Identification
Our service harnesses the power of mass spectrometry, the sole method capable of precisely pinpointing the exact citrullination sites on proteins. This precision offers invaluable insights into the functional ramifications of this post-translational modification.
Exceptional Sensitivity
Utilizing state-of-the-art mass spectrometry instruments, we achieve remarkable sensitivity, enabling the detection of even the most minute quantities of citrullinated proteins. This heightened sensitivity is particularly advantageous for identifying low-abundance citrullinated proteins.
Comprehensive Understanding
Our service offers a comprehensive overview of protein citrullination, spanning from meticulous sample preparation to thorough data analysis. Researchers can delve deep into the implications of citrullination across various biological processes and diseases, gaining a holistic understanding of its role.
Quantitative Proficiency
We employ quantitative proteomics techniques such as SILAC and DIA (Data-Independent Acquisition) proteomics to quantify citrullination levels accurately. This quantitative analysis facilitates comparisons across diverse biological conditions, further enhancing the depth of our insights.
Versatile and Customized Enrichment Solutions
Tailoring our enrichment strategies to the specific requirements of each project ensures a smooth transition to subsequent mass spectrometry analysis. Whether your focus is on global deubiquitination patterns or specific proteins and pathways, we can customize enrichment strategies to meet your research objectives.
References
- Daniel M. Lewallen, Kevin L. Bicker, Venkataraman Subramanian, Kathleen W. Clancy, Daniel J. Slade, Julianne Martell, Christina J. Dreyton, Jeremy Sokolove, Eranthie Weerapana, and Paul R. Thompson Chemical Proteomic Platform To Identify Citrullinated Proteins. ACS Chemical Biology 2015
- Erdem Cicek, Gerald Monard, and Fethiye Aylin Sungur Molecular. Mechanism of Protein Arginine Deiminase 2: A Study Involving Multiple Microsecond Long Molecular Dynamics Simulations. Biochemistry 2022
- Yatao Shi, Zihui Li, Bin Wang, Xudong Shi, Hui Ye, Daniel G. Delafield, Langlang Lv, Zhengqing Ye, Zhengwei Chen, Fengfei Ma, and Lingjun Li Enabling Global Analysis of Protein Citrullination via Biotin Thiol Tag-Assisted Mass Spectrometry Analytical Chemistry 2022 94 (51), 17895-17903
- Ronak Tilvawala1, Paul R. Thompson Peptidyl Arginine Deiminases: Detection and Functional Analysis of Protein Citrullination Curr Opin Struct Biol. 2019
Our products and services are for research use only.

