New quantitative mass spectrometry (MS) and bioinformatics workflows are maturing in both label-free and label multiplexing technologies, providing a solid foundation for PTM analysis. Sequential Window Data Independent Acquisition (DIA)/Acquisition of all Theoretical fragment ions (SWATH) is one of these methods and has been emerging as a promising approach due to its re-mining capability. We are now providing a high-throughput, label-free PTM quantification approach, the comprehensive DIA/SWATH for PTM quantitative analysis.
Overview of DIA/SWATH quantification
With the increased data acquisition rates of MS instruments today, many large-scale proteomics studies using MS techniques rely on data acquisition (DDA). A large number of peptide-spectrum matches are generated, but missing values in individual DDA experiments can lead to a degree of incompleteness in large data sets due to the stochastic nature of the technology. DIA methods like SWATH systematically fragment all precursor ions in a user-defined retention time vs. precursor ion mass to charge (m/z) window, which can overcome to some extent the missing values in quantitative studies. The recent popularity of DIA-based methods is mainly due to their good performance on high-resolution, precision mass instruments including accurate quantification over a dynamic range and highly consistent detection of analytes across sample cohorts.
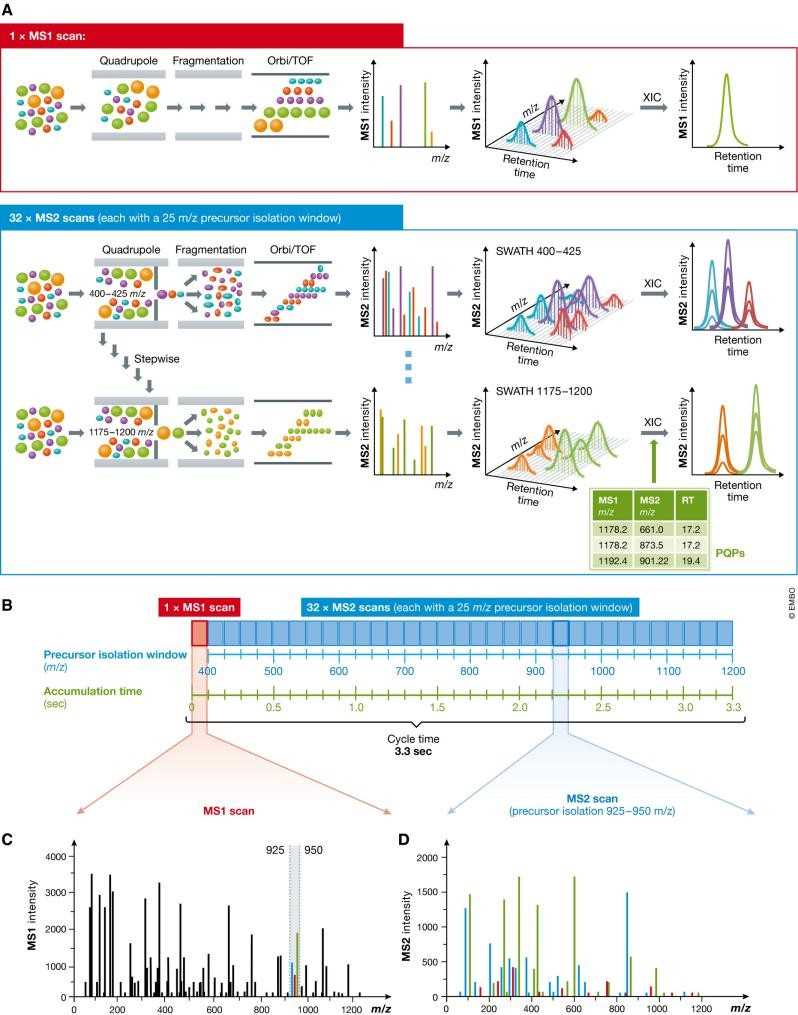 Fig. 1 Principle of sequentially windowed data-independent acquisition in SWATH-MS. (Ludwig, Christina, et al., 2018)
Fig. 1 Principle of sequentially windowed data-independent acquisition in SWATH-MS. (Ludwig, Christina, et al., 2018)
Key features of SWATH-MS
- Accurate quantification over a dynamic range.
- Highly consistent detection of analytes across sample cohorts.
- High sensitivity, enabling detection of modified peptides at lower abundance.
- Suitable for projects that entail a large number of samples.
Sequential windowed acquisition of all theoretical fragment ion mass spectrometry (SWATH-MS) proteomics analysis
The SWATH-MS method enables a more in-depth recording of the peptide contents of samples as well as their modifications. Based on the pre-generated spectral libraries and the analysis of the precursor ions, the SWATH-MS data can be targeted for querying to identify and quantify peptides in the samples. We have experienced experts and improved analysis workflow, allowing for low abundance sub-proteome characterization, especially in PTM analysis, such as glycosylation, acetylation, and succinylation. We are dedicated to helping researchers and professionals study complex biological samples in high reproducibility, speed, and resolution manner.
Workflow of our service
- Experimental design and consultation.
- Protein digestion and sample preparation for MS analysis.
- Peptide enrichment.
- LC-MS/MS and SWATH-MS analysis.
- Spectral library and SWATH-MS assay construction.
- Data processing and OpenSwath software analysis, including raw SWATH-MS file conversion, library analysis, and identification of peptide and their modifications.
- Bioinformatics analysis.
Sample requirements
- Suitable for analyzing various types of samples, such as tissue, cell, and biological fluids.
- Sample shipping: sufficient amount of dry ice for shipping, or consult our technical staff before sending samples.
- Before the formal experiment, we will always test the samples you provide.
- If you want to know specific sample requirements, please feel free to contact us.
Related services
For clients wishing to quantify protein PTMs by MS analysis, Creative Proteomics now offers a range of labeled and unlabeled proteomic-based methods and services. We are committed to developing a more intuitive and streamlined process for researchers and professionals in the field of protein PTM analysis. Please feel free to contact us for more details on our services.
References
- Ludwig, Christina, et al. "Data-independent acquisition-based SWATH-MS for quantitative proteomics: a tutorial." Molecular systems biology 14.8 (2018): e8126.
- Sauer, Christopher S., et al. "Developing mass spectrometry for the quantitative analysis of neuropeptides." Expert Review of Proteomics 18.7 (2021): 607-621.
Services You May Be Interested In
Our products and services are for research use only.