S-glutathionylation is a reversible post-translational modification involving the addition of glutathione (GSH) to cysteine residues of proteins and plays a pivotal role in cellular redox regulation. This process is integral to maintaining cellular homeostasis and has garnered significant attention in the field of molecular biology. Understanding the intricacies of S-glutathionylation is crucial for unraveling its implications for health and disease.
Analytical Methods for S-Glutathionylation
- Mass Spectrometry. Mass spectrometry stands at the forefront of S-glutathionylation analysis, offering unparalleled sensitivity and resolution. High-resolution MS techniques, such as tandem mass spectrometry (MS/MS), enable precise identification of modified peptides. Advanced labeling strategies, including isotope-coded affinity tags (ICAT) and iodoacetamide-based methods, enhance detection efficiency.
- Western Blotting. Western blotting remains a reliable technique for probing S-Glutathionylation. Utilizing antibodies specific to glutathione-conjugated proteins, this method allows for semi-quantitative assessment. Improved blotting strategies, such as non-reducing conditions, enhance the detection of S-glutathionylation, providing insights into protein oxidation states.
- Redox Proteomics. Redox proteomics employs innovative approaches to capture and identify S-glutathionylated proteins. Techniques like biotin switch assays and redox difference gel electrophoresis (redox DIGE) facilitate the global profiling of S-glutathionylation events. This holistic analysis offers a comprehensive view of redox-regulated protein networks.
Applications of S-Glutathionylation Analysis
- Disease Pathogenesis. S-glutathionylation has emerged as a key player in various pathological conditions. Analyzing S-glutathionylated proteins in diseases like cancer, neurodegenerative disorders, and cardiovascular diseases unveils potential therapeutic targets. Understanding the molecular mechanisms underlying S-glutathionylation aids in developing targeted interventions.
- Drug Development. The pharmaceutical industry leverages S-glutathionylation analysis to assess the impact of drugs on cellular redox status. Investigating drug-induced changes in S-glutathionylation patterns provides valuable insights into drug efficacy and toxicity. This knowledge is pivotal in refining drug development pipelines and ensuring the safety of therapeutic agents.
- Environmental Stress Response. Cellular adaption mechanisms are clarified by researching S-glutathionylation in response to external stresses including oxidative stress and pollution. Examining the redox state of proteins under stress advances our knowledge of how resilient cells are to environmental harm and opens up new possibilities for strategy development.
Other Important Aspects in S-Glutathionylation Analysis
- Quantitative Assessment. Understanding the biological importance of S-glutathionylation events requires accurate measurement. Enhancing S-Glutathionylation analysis precision is possible through the development of robust quantitative techniques like as label-free quantification and stable isotope labeling. This quantitative data is essential for understanding how redox signaling is dynamic.
- Functional Consequences. It is critical to clarify the functional effects of S-glutathionylation on protein activity. A comprehensive understanding of how this alteration affects protein function can be obtained by combining functional experiments with S-glutathionylation studies. This realization is crucial to understanding the knock-on effects on signaling pathways and cellular functions.
Advantages of Our Services

An essential tool for studying redox biology is S-glutathionylation analysis. Exploiting cutting-edge analytical techniques and using this information in a variety of domains, such as medication creation and illness research, creates new opportunities for scientific investigation. As we continue to delve deeper into the complexities of S-Glutathionylation, the potential for therapeutic interventions and a deeper understanding of cellular redox regulation becomes increasingly apparent.
Oxidative stress-induced FABP5 S-glutathionylation protects against acute lung injury by suppressing inflammation in macrophages
Journal: Nature Communications
Published: 2021
Background
Under conditions of hyperoxia and LPS-induced acute lung injury, protein S-glutathionylation modification is increased and the expression of the corresponding deglutathionase Grx1 is decreased, especially FABP5 is susceptible to glutathione modification at the cysteine 127 site under oxidative conditions thereby promoting its fatty acid binding and nuclear localization, and facilitating the interaction of FABP5 with PPARβ/δ and activation of PPARβ/δ downstream target genes to inhibit LPS-induced macrophage inflammation. In this paper, we will specifically elucidate the mechanism of FABP5 cysteine glutathione modification on the regulation of the macrophage inflammatory process during the case process of acute lung injury.
Results
C57 mice were randomly grouped and exposed to normoxic or hyperoxic conditions, respectively, and the ratio of reduced glutathione GSH/oxidized glutathione was decreased in hyperoxia, and high expression of GSH synthase (Gss) was found. The results suggest an association between redox regulation and hyperoxic acute kidney injury. Analysis of key enzymes involved in glutathionylation revealed a significant decrease in the expression level of the deglutathione enzyme Grx1, while the expression levels of Grx2 and the glutathione-S-transferase Gsto 1-1 remained unchanged. And the enzyme activity of Grx1 was reduced under hyperoxia exposure conditions. These results suggest that Grx1 is involved in the pathologic process of lung injury induced by hyperoxia, and the lung injury condition of Grx1 knockout mice was significantly improved under hyperoxia exposure. Moreover, granulocytes, and cytokines were significantly reduced under this condition. And the overall protein S-glutathionylation level was increased. In Grx1 knockout mice, protein S-glutathionylation levels were solidly elevated after hyperoxia exposure compared to controls. Thus, these results suggest the importance of Grx1 and S-glutathionylation in hyperoxia-induced lung injury. We suggest that the reduction in Grx1 expression levels and activity in mouse lung tissue plays a protective role against oxidative stress (Figure 1).
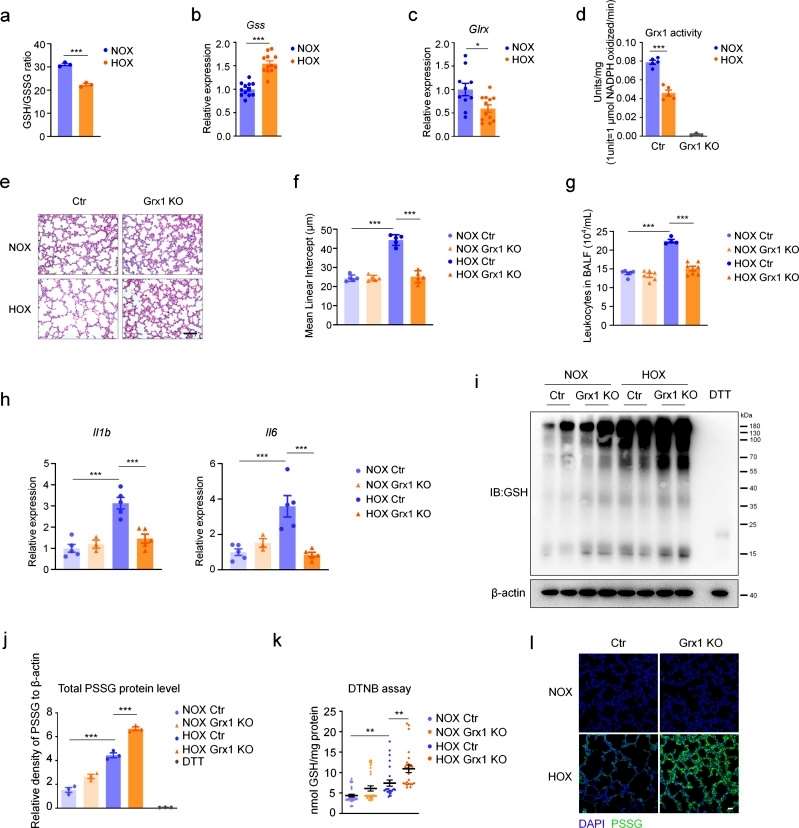 Figure 1
Figure 1
The authors next explored which cell type is most sensitive to oxidative stress during the pathologic process of acute lung injury by detecting intracellular ROS levels with a fluorescent probe, and found that total ROS levels were significantly increased and the intracellular GSH/GSSG ratio was significantly decreased in macrophages in the hyperoxia-exposed group compared with the control group, and that Grx1 levels were significantly decreased in hyperoxia-exposed alveolar macrophages (AMs). Moreover, Grx1 deprivation significantly increased protein S-glutathionylation levels in AMs when exposed to hyperoxic conditions. It was next verified whether Grx1 plays an important role in acute lung injury by regulating S-glutathionylation levels in macrophages. Conditional macrophage-specific knockout Grx1 and wild-type mice showed a significant increase in the level of ROS in macrophages under LPS stimulation conditions, and histologic analysis revealed that the inflammatory condition was significantly suppressed in Grx1 knockout mice. And this protective effect was further evidenced by the reduction of granulocytes in alveolar lavage fluid. The expression levels of pro-inflammatory cytokines were also significantly reduced, and the results overall suggest that Grx1-regulated S-glutathionylation in macrophages plays a protective role in acute lung injury (Figure 2).
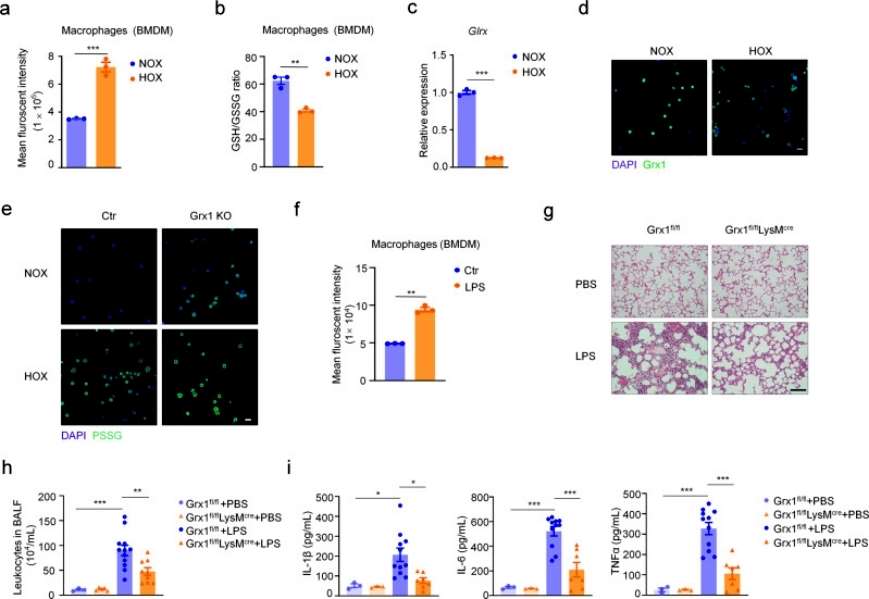 Figure 2
Figure 2
To identify Grx1-specific molecular targets and pathways susceptible to redox-dependent regulation in acute lung injury pathology. S-glutathionylation site-specific analysis was performed within the macrophage proteome using a quantitative redox proteomics approach. glutathionylated proteins were enriched and quantified after BMDM exposure to 200 μM H2O2 treatment for 15 min, and GO analysis of S-glutathionylated proteins revealed that the main enriched distributions were found in the major cellular, metabolic, and single-species processes. Among the top five modified proteins of metabolic processes, FABP5 was expressed at significantly higher levels in macrophages than other proteins. Overall, S-glutathionylation of FABP5 is involved in the regulation of macrophage acute lung injury pathological processes (Figure 3).
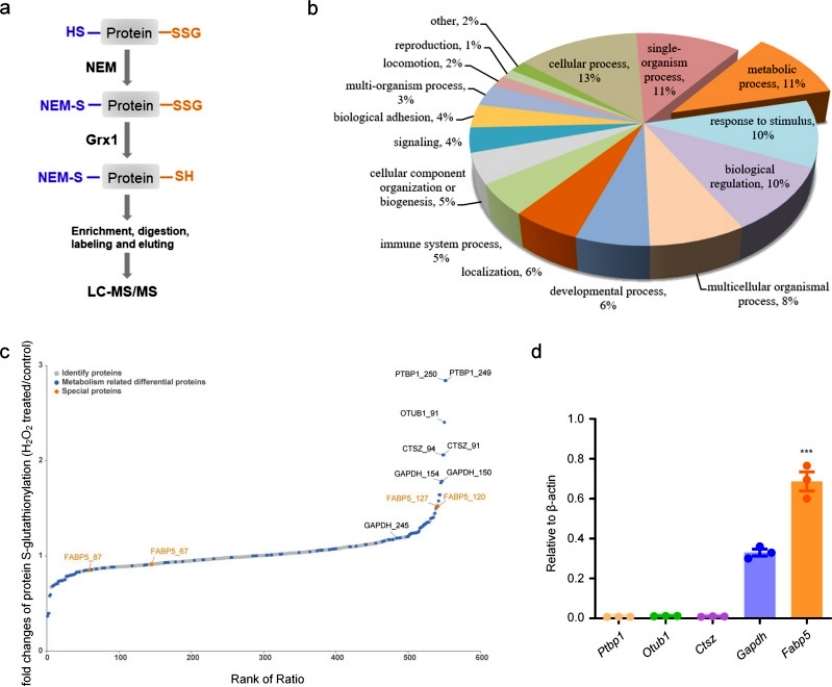 Figure 3
Figure 3
Conclusion
The authors showed that ROS promoted S-glutathionylation of FABP5 in macrophages under oxidative conditions. S-glutathionylation enhanced fatty acid binding and nuclear accumulation of FABP5, activated PPARβ / δ, and inhibited LPS-induced macrophage inflammation. These data confirm Grx1 as a target for anti-inflammatory drug development and suggest that S-glutathionylation of FABP5 acts as an anti-inflammatory mediator in macrophages during acute lung injury. The findings reveal a mechanism by which macrophages prevent self-hyperactivation in response to oxidative stress.
Reference
- Guo Y, Liu Y, Zhao S, et al., Oxidative stress-induced FABP5 S-glutathionylation protects against acute lung injury by suppressing inflammation in macrophages. Nat Commun. 2021 Dec 7;12(1):7094.

