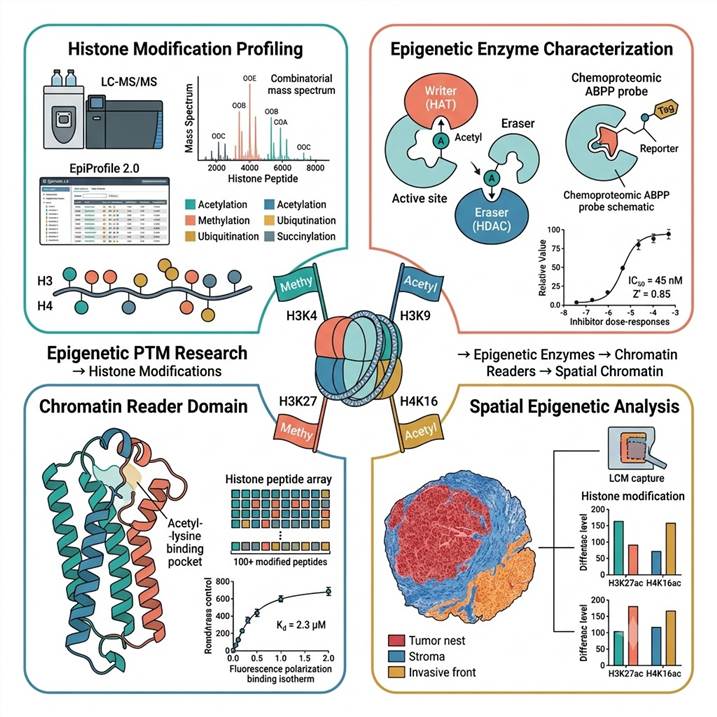
Epigenetic PTM Research — From Histone Modification Maps to Epigenetic Drug Targets
Human chromatin biology is controlled by an estimated 800+ proteins that write, read, and erase histone post-translational modifications. Understanding this network requires knowing which modifications co-occur on the same histone molecule, which enzymes control them, which reader domains interpret each pattern, and how these interactions change in disease. Our integrated MS platform addresses all four dimensions:
- Histone modification mapping: LC-MS/MS quantification of acetylation, mono-/di-/trimethylation, ubiquitination, succinylation, hydroxyisobutyrylation, and acyl modifications at site-specific resolution across H1–H4. Both site occupancy and combinatorial peptidoform distributions reported.
- Epigenetic enzyme characterization: Activity-based assays for HATs (p300/CBP, PCAF/GCN5, MYST), HDACs (class I, IIb, IV), HMTs (G9a, EZH2, SETD2, PRMTs), KDMs (LSD1, KDM4–6), and histone kinases. Chemoproteomic ABPP for inhibitor target engagement and selectivity.
- Chromatin reader domain analysis: Histone peptide arrays (100+ modified peptides) to map modification specificity of bromodomains, chromodomains, PHD fingers, Tudor, MBT, YEATS, and BAH domains. Quantitative Kd by FP and ITC.
- Spatial chromatin analysis: Tissue-level histone modification profiling by LCM-MS, revealing intratumoral and intercellular chromatin heterogeneity invisible to bulk analysis.
Every dataset includes modification crosstalk analysis, enzyme-substrate relationship mapping, and reader domain specificity annotation — connecting modification lists to mechanistic insight.
| Service Module |
Technology |
Key Deliverable |
Research Application |
| Histone PTM Profiling |
Bottom-up LC-MS/MS (DIA/DDA); EpiProfile 2.0 + Skyline |
Site-specific modification occupancy; combinatorial peptidoform distributions across H1–H4 |
Chromatin state mapping, disease vs. normal comparison, drug treatment response |
| Single-Cell Histone PTM Analysis |
cellenONE + timsTOF Ultra DIA; sc-hPTM workflow |
67+ histone peptidoforms per single cell; cellular heterogeneity resolved |
Cell-to-cell chromatin heterogeneity, rare subpopulation identification |
| Epigenetic Writer/Eraser Enzyme Assays |
Fluorogenic HAT/HDAC/HMT/KDM activity assays; chemoproteomic ABPP |
IC₅₀ values for inhibitors; enzyme substrate specificity profiles; target engagement data |
Epigenetic drug discovery, inhibitor SAR, enzyme mechanism studies |
| Chromatin Reader Domain Analysis |
Histone peptide arrays; FP/ITC binding quantification |
Modification specificity matrix; Kd values for reader–histone peptide interactions |
Reader domain drug targeting, chromatin recruitment mechanism studies |
| Spatial Histone Modification Profiling |
Spatially resolved MS with laser capture microdissection |
Histone modification maps across tissue regions; cell-type-specific chromatin signatures |
Tumor heterogeneity, tissue microenvironment, developmental gradients |
| Epigenetic Multi-Omics Integration |
Proteomics + histone PTM + transcriptomics co-analysis |
Integrated chromatin state → gene expression correlation; pathway-level epigenetic regulation maps |
Systems epigenetics, mechanism-of-action studies, biomarker discovery |
Epigenetic PTM Analysis Platforms
Histone Modification Profiling by LC-MS/MS
Bottom-up LC-MS/MS with EpiProfile 2.0 analysis circumvents antibody limitations — quantifying acetylation, mono-/di-/trimethylation, ubiquitination, succinylation, and hydroxyisobutyrylation across all core histones (H1–H4). Propionyl derivatization standardizes tryptic digestion; DIA/DDA acquisition captures full peptidoform data. We report site-level occupancy and combinatorial peptidoform distributions (CV < 15%).
Single-Cell Histone PTM Analysis (sc-hPTM)
cellenONE single-cell dispensing + Bruker timsTOF Ultra DIA with diaPASEF quantifies 67+ histone peptidoforms per individual cell. Data completeness exceeds 99.9% (<0.1% missing values); signal-to-noise averages 9.5-fold over blank. Reveals cell-to-cell chromatin heterogeneity — rare subpopulations and drug-tolerant persister states invisible to bulk analysis.
Epigenetic Enzyme Activity & Inhibitor Profiling
Biochemical and chemoproteomic assays for writers (HATs, HMTs, kinases), erasers (HDACs, KDMs, phosphatases), and their regulators. Fluorogenic substrate readout in 384-well format for IC₅₀ determination. ABPP probes (SAHA-BPyne for HDACs) map target engagement against endogenous enzymes in native complexes. Integrates with HDAC/HAT assays and PTM enzyme inhibitor screening.
Chromatin Reader Domain Characterization
Histone peptide arrays (100+ modified peptides) map modification specificity of bromodomains, chromodomains, PHD fingers, Tudor, MBT, YEATS, and BAH domains. Quantitative Kd determination by FP and ITC. Competitive displacement assays for reader domain inhibitor discovery.
Spatial Histone Modification Analysis
Laser capture microdissection (LCM) combined with LC-MS/MS histone PTM profiling generates spatially resolved chromatin modification maps across tissue regions. Reveals tumor vs. stroma, proliferating vs. quiescent zone, and invasive front chromatin signatures. Integrates with spatial histone modification profiling.
Applications in Epigenetic Research and Drug Discovery
Epigenetic Drug Discovery
- HDAC inhibitor selectivity profiling across class I/IIb/IV isoforms with cellular histone acetylation PD readout
- HAT inhibitor screening with p300/CBP and PCAF/GCN5 specificity panels
- HMT inhibitor characterization (EZH2, G9a, DOT1L, PRMT1–9) with SAM-competitive mechanism determination
- Bromodomain inhibitor screening (BRD4-BD1/BD2, CBP/p300-BRD, etc.) with FP-based competitive displacement
Chromatin State Mapping
- Comprehensive histone modification surveys across treatment conditions, time points, or disease states
- Combinatorial peptidoform analysis revealing co-occurring modifications on the same histone molecule
- Differential modification analysis with statistical thresholds and biological pathway annotation
- Integration with acetylomics for non-histone acetylation context
Epigenetic Reader Domain Drug Targeting
- Modification specificity mapping for novel reader domains using histone peptide arrays
- Quantitative Kd determination for reader–histone peptide interactions by FP and ITC
- Competitive inhibitor screening against reader domains with modification-specific peptide probes
- Selectivity profiling across related reader domain family members
Disease Mechanism Studies
- Histone modification landscape comparison between disease and normal tissue
- Epigenetic enzyme expression and activity correlation with chromatin state changes
- Mutation-specific chromatin state analysis for oncohistone mutations (H3K27M, H3G34R/V, H3K36M)
- Time-resolved histone modification dynamics during cellular differentiation or drug treatment
Epigenetic Biomarker Discovery
- Histone modification signatures as biomarkers for drug response or disease progression
- Single-cell histone PTM heterogeneity as a predictor of drug tolerance or resistance
- Circulating histone modification analysis from plasma or biofluids
- Epigenetic multi-omics integration — histone PTM + proteomics + transcriptomics
Tissue-Level Epigenetic Heterogeneity
- Spatially resolved histone modification maps across tumor regions by LCM-MS
- Cell-type-specific chromatin signatures from complex tissues
- Spatial epigenetic profiling integrated with spatial PTM profiling for multi-mark analysis
- Microenvironment-driven chromatin state characterization
Epigenetic PTM Research Workflow
1. Experimental Design
- Define research objective: histone modification profiling, enzyme activity characterization, reader domain analysis, or spatial chromatin mapping
- Sample type and quantity requirements determined: cells (10⁶–10⁷), tissue (10–50 mg), or recombinant proteins
- Histone extraction protocol selection: acid extraction (core histones), salt extraction (chromatin-bound proteins), or native nucleosome preparation
- Modification panel design: targeted marks of interest or global histone PTM survey
2. Sample Preparation
- Histone enrichment: acid extraction with HDAC/protease/phosphatase inhibitor cocktails
- Propionyl derivatization to block unmodified lysines and control tryptic digestion
- For sc-hPTM: single-cell dispensing by cellenONE into 384-well plates with one-pot derivatization and digestion
- For enzyme assays: recombinant enzyme QC, substrate preparation, inhibitor dilution series
3. LC-MS/MS Data Acquisition
- Bulk histone PTM: DDA or DIA on Orbitrap Exploris 480 or timsTOF Pro 2
- sc-hPTM: DIA with diaPASEF on Bruker timsTOF Ultra for single-cell sensitivity
- Targeted PRM for low-abundance modification site verification
- Reader domain assays: FP and ITC on plate readers and microcalorimeters
4. Data Processing
- Histone PTM: EpiProfile 2.0 for histone peptidoform identification; Skyline for manual verification
- Site-level modification quantification with relative abundance reporting (% of total histone)
- Combinatorial modification analysis: co-occurring PTM pattern enumeration and statistical enrichment
- Enzyme assay data: IC₅₀ by four-parameter logistic regression; Z′ factor QC
5. Epigenetic Network Integration
- Modification crosstalk analysis: identifying coordinated and mutually exclusive modification patterns
- Enzyme-substrate relationship mapping: which modifications are controlled by which enzyme in your system
- Reader domain annotation: which detected modifications are recognized by which reader domains
- Multi-omics integration: histone PTM + proteomics + transcriptomics co-analysis for systems-level chromatin biology
6. Reporting and Data Delivery
- Complete histone modification quantification tables with site and peptidoform resolution
- Combinatorial modification network diagrams showing coordinated modification patterns
- Volcano plots and heat maps for differential modification analysis across conditions
- Raw data files (mzML, EpiProfile output), publication-ready figures, and methods documentation
Why Choose Our Epigenetic PTM Research Platform
Full Epigenetic PTM Coverage — Not Just Histone Marks
- Writer, reader, and eraser analysis in a single provider — no need to split your project across multiple CROs
- Histone modification profiling + enzyme activity + reader domain binding + spatial analysis, all under one quality system
- Data from all modules share consistent sample preparation, instrumentation, and bioinformatics pipelines
Combinatorial Modification Analysis — The Missing Dimension
- Antibody-based methods report one mark at a time; we report which marks coexist on the same histone molecule
- Combinatorial peptidoform analysis reveals modification crosstalk — coordinated patterns invisible to single-mark detection
- Modification co-occurrence data directly informs reader domain binding predictions and chromatin state models
From Single Cell to Tissue — Scale Matched to Biology
- Single-cell histone PTM analysis reveals epigenetic heterogeneity — identifying rare chromatin states that drive resistance
- Bulk profiling provides population-average modification landscapes for statistically powered condition comparisons
- Spatial histone analysis maps chromatin states across tissue architecture — connecting epigenetics to histology
Case Study — Single-Cell Histone PTM Profiling by LC-MS/MS Reveals Chromatin Heterogeneity
Cutler, Corveleyn, Ctortecka, and colleagues developed sc-hPTM — a robust mass spectrometry platform for profiling histone post-translational modifications in single cells — demonstrating how single-cell chromatin analysis reveals epigenetic heterogeneity that bulk methods cannot detect (Cutler et al., 2025, Nature Communications, CC BY 4.0).
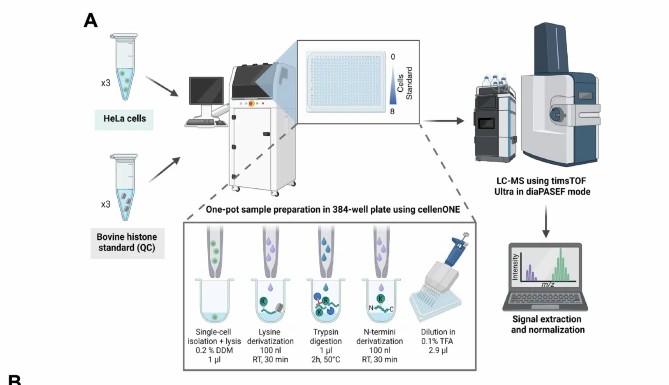
Background: Histone post-translational modifications are central to chromatin regulation — they control DNA accessibility, transcriptional activity, replication timing, and DNA repair. The standard method for histone modification analysis, bottom-up LC-MS/MS on bulk cell populations, quantifies modification states averaged across millions of cells. This averaging obscures the cellular heterogeneity that is increasingly recognized as a driver of drug resistance, tumor evolution, and developmental cell-fate decisions. The team set out to develop a mass spectrometry method capable of robustly profiling histone PTMs in single cells — overcoming the sensitivity and data completeness challenges that had prevented single-cell chromatin analysis at the modification level.
Methods: The sc-hPTM platform integrated four innovations: (1) cellenONE single-cell dispensing into 384-well plates for precise single-cell isolation; (2) one-pot propionyl derivatization and trypsin digestion optimized for picogram-level histone input; (3) data-independent acquisition with diaPASEF on a Bruker timsTOF Ultra mass spectrometer, providing ion mobility separation and high sensitivity; and (4) EpiProfile 2.0-based data analysis for histone peptidoform identification and label-free quantification. The platform was validated using HeLa cells treated with sodium butyrate (a broad-spectrum HDAC inhibitor) to induce histone hyperacetylation, and the single-cell data were compared with matched bulk analyses. Key metrics included peptidoform coverage per cell, quantitative reproducibility, and the ability to distinguish treated from untreated cells at the single-cell level.
Results: The sc-hPTM platform identified 67 histone peptidoforms across H1, H2A, H2B, H3, and H4 in single HeLa cells, covering acetylation, mono-/di-/trimethylation, ubiquitination, succinylation, and hydroxyisobutyrylation. Data completeness exceeded 99.9% (<0.1% missing values), and signal-to-noise ratios averaged 9.5-fold over blank controls. Sodium butyrate treatment (5 mM, 24 h) induced dramatic histone hyperacetylation — single-cell analysis revealed that treated and untreated cells formed distinct clusters based on their histone modification profiles, with clear separation driven by H3K9ac, H3K14ac, H4K5ac, H4K12ac, and H4K16ac. Crucially, sc-hPTM identified a subpopulation of sodium butyrate-treated cells (~15%) that showed only partial hyperacetylation — a heterogeneous response invisible to bulk analysis, where all treated cells appeared uniformly hyperacetylated. The platform also revealed differential co-regulation patterns: for example, H4K16ac and H4K20me3 showed mutually exclusive patterns in single cells, a relationship masked by population averaging.
Significance for Epigenetic PTM Research: This study demonstrates that mass spectrometry-based single-cell histone PTM profiling is now a practical tool for chromatin biology — enabling the detection of epigenetic heterogeneity that bulk methods and antibody-based approaches cannot resolve. For epigenetic drug discovery programs, the sc-hPTM platform provides a direct readout of cellular heterogeneity in drug response — identifying subpopulations with incomplete target engagement or alternative chromatin states that may drive drug tolerance. For basic chromatin biology, the platform enables the study of epigenetic cell-to-cell variation during development, differentiation, and disease progression. The integration of sc-hPTM with the broader epigenetic PTM research platform — histone modification profiling at bulk and single-cell resolution, epigenetic enzyme activity assays, reader domain characterization, and spatial chromatin analysis — enables a comprehensive approach to chromatin biology: from modification identification through functional validation to cellular-level mechanism.
Reference: Mass spectrometry-based profiling of single-cell histone post-translational modifications to dissect chromatin heterogeneity. Cutler A, Corveleyn L, Ctortecka C, LiCausi N, Cantlon J, Vaca Jacome AS, Deforce D, Vijg J, Dhaenens M, Papanastasiou M, Carr SA, Sidoli S. Nature Communications. 2025;16:11100. (CC BY 4.0)
Related Services
Our epigenetic PTM research platform integrates with the following histone modification, epigenetic enzyme, and spatial chromatin analysis services:
Frequently Asked Questions
What histone modifications can you detect and quantify?
We quantify acetylation (Kac), mono-/di-/trimethylation (Kme1/me2/me3), ubiquitination, succinylation, and hydroxyisobutyrylation across all core histones (H1, H2A, H2B, H3, H4) and linker histone H1. In a typical bulk histone PTM profiling experiment, we report 60–80 histone peptidoforms covering the major regulatory marks including H3K4me1/me2/me3, H3K9me1/me2/me3, H3K9ac, H3K14ac, H3K18ac, H3K23ac, H3K27me1/me2/me3, H3K27ac, H3K36me1/me2/me3, H3K79me1/me2, H4K5ac, H4K8ac, H4K12ac, H4K16ac, H4K20me1/me2/me3, H2AK5ac, H2AK9ac, and many combinatorial peptidoforms. Our single-cell platform identifies 67+ peptidoforms per individual cell. Custom modification panels and targeted PRM assays are available for specific marks of interest.
How does LC-MS/MS histone analysis compare to antibody-based methods (ChIP-seq, Western blot)?
LC-MS/MS offers three key advantages over antibody-based methods: (1) Antibody independence — no issues with antibody specificity, lot-to-lot variability, or epitope occlusion by neighboring modifications. Every modified peptide is identified by its mass and fragmentation pattern, not by antibody binding. (2) Combinatorial modification analysis — MS detects which modifications coexist on the same histone peptide (e.g., H3K9me2 + H3K14ac on the same molecule), which antibodies cannot determine. (3) Discovery-mode capability — MS detects modifications without prior hypothesis, enabling identification of unexpected marks or combinatorial patterns. The trade-off is that MS does not provide genomic location information (where the modification is along the genome), which ChIP-seq does provide. For comprehensive histone modification profiling and combinatorial analysis, LC-MS/MS is the gold standard; ChIP-seq is complementary for locus-specific analysis.
Can you analyze histone modifications at single-cell resolution?
Yes — our sc-hPTM platform combines cellenONE single-cell dispensing with Bruker timsTOF Ultra DIA mass spectrometry to quantify 67+ histone peptidoforms per single cell. The workflow achieves >99.9% data completeness and robust separation of treated vs. untreated cells based on their histone modification profiles. Applications include: identifying drug-tolerant persister subpopulations with distinct chromatin states, characterizing epigenetic heterogeneity during differentiation or disease progression, and detecting rare cells with aberrant histone modification patterns. The single-cell platform is complementary to our bulk histone PTM profiling service — we recommend bulk profiling for initial chromatin state characterization and single-cell analysis for heterogeneity-focused questions.
What epigenetic enzyme classes can you assay for inhibitor screening?
We provide activity-based assays for histone acetyltransferases (p300/CBP, PCAF/GCN5, MYST family including Tip60 and MOF), histone deacetylases (HDAC1/2/3/6/8/10/11 with class I/IIb/IV selectivity panels), histone methyltransferases (G9a, GLP, EZH2, SETD2, DOT1L, PRMT1–9, NSD1–3, SUV39H1/2, SETDB1), lysine demethylases (LSD1/KDM1A, KDM4A–D, KDM5A–D, KDM6A/B), and histone kinases (Haspin, Aurora B, MSK1/2). Biochemical assays use fluorogenic or luminescence readout in 384-well format. Chemoproteomic ABPP profiling is available for HDACs (SAHA-BPyne probe) and can be developed for other enzyme classes. Custom assay development is available for novel epigenetic enzyme targets.
How do you handle the challenge of combinatorial histone modification complexity?
Histone modification analysis is complicated by the vast combinatorial space — with dozens of modification sites on each histone, the number of possible peptidoforms is enormous. We address this through three strategies: (1) Propionyl derivatization to standardize tryptic digestion of hyper-modified histones, producing peptides of predictable length amenable to LC-MS/MS. (2) EpiProfile 2.0 software, which uses a validated library of histone peptide retention times and fragmentation patterns to identify and quantify peptidoforms, including isobaric forms that differ only in modification position. (3) Data-independent acquisition (DIA) to capture fragment ion evidence for all peptidoforms simultaneously, enabling retrospective re-analysis if new modification hypotheses arise. Our reports include both site-level quantification (total modification at each residue, summed across all peptidoforms) and peptidoform-level quantification (relative abundance of each combinatorial form), enabling both reductionist and systems-level analysis of your chromatin modification data.
What sample types and quantities are required for histone PTM analysis?
For bulk histone PTM profiling: 1–10 million cells or 10–50 mg tissue per condition. Histones are acid-extracted, which enriches for core histones (H2A, H2B, H3, H4) and linker histone H1 while removing most non-histone proteins. For single-cell histone PTM analysis: intact, viable cells are required for cellenONE dispensing. For epigenetic enzyme activity assays: recombinant enzymes or nuclear extracts with the target enzyme activity confirmed. For reader domain analysis: purified recombinant reader domain protein (typically 100 µg minimum). For spatial histone analysis: fresh-frozen or FFPE tissue sections — FFPE-compatible protocols are available for histone extraction from archived specimens. Detailed sample preparation guidelines are provided for each module.
References
- Mass spectrometry-based profiling of single-cell histone post-translational modifications to dissect chromatin heterogeneity. Cutler A, Corveleyn L, Ctortecka C, LiCausi N, Cantlon J, Vaca Jacome AS, Deforce D, Vijg J, Dhaenens M, Papanastasiou M, Carr SA, Sidoli S. Nature Communications. 2025;16:11100.
- Improved mass spectrometry-based methods reveal abundant propionylation and tissue-specific histone propionylation profiles. Vai A, Noberini R, Ghirardi C, Rodrigues de Paula D, Carminati M, Pallavi R, Araújo N, Varga-Weisz P, Bonaldi T. Molecular & Cellular Proteomics. 2024;23(7):100799.
- Cancer epigenetics: from laboratory studies and clinical trials to precision medicine. Yu X, Zhao H, Wang R, Chen Y, Ouyang X, Li W, Sun Y, Peng A. Cell Death Discovery. 2024;10:28.
This service is provided for research use only (RUO). Not for diagnostic or clinical applications.