
Introduction to Histone Biology and Epigenetics
Histones are highly conserved nuclear proteins. They serve as the principal protein components of chromatin. Each histone molecule contributes to the structural organization of the genome by forming nucleosomes, which compact and regulate access to DNA. The nucleosome core comprises an octamer of four core histones (H2A, H2B, H3, and H4) around which approximately 147 base pairs of DNA are wrapped.
Beyond structural roles, histones participate in the regulation of gene expression through a diverse array of post-translational modifications (PTMs). These modifications include acetylation, methylation, phosphorylation, ubiquitination, and novel acylations. They occur primarily on the flexible N-terminal tails of histones and modulate chromatin accessibility and transcription factor binding.
The functional impact of histone PTMs is mediated by regulatory enzymes classified as writers, erasers, and readers. These enzymes frequently rely on cellular metabolites, positioning histone PTMs as sensitive indicators of the metabolic state. The ensemble of histone modifications, often referred to as the "histone code," orchestrates transcriptional outcomes and chromatin remodeling.
Overview of Histone Isolation Methods
Quantitative analysis of histone PTMs requires high-purity histone preparations. Histone isolation methods must preserve PTM integrity while ensuring compatibility with downstream analytical platforms such as mass spectrometry (MS).
Total vs. Core Histone Isolation
Total histone isolation involves the extraction of all nuclear histones, including linker histone H1. In contrast, core histone isolation selectively retrieves H2A, H2B, H3, and H4. The decision depends on the experimental goal. Core histones are typically enriched for PTMs and are preferred for PTM profiling. Total histones may be suitable for stoichiometric or compositional studies.
Key Considerations Before Starting Histone Extraction
Histone extraction protocols must balance yield, purity, and modification preservation. Important factors include:
- Biological source (cell line, tissue type, or organism)
- Sample input quantity
- Histone variant abundance
- PTM sensitivity to processing steps
How Cell Type and Sample Size Affect Your Strategy
Different cell types exhibit variable chromatin states and histone expression levels. For example, proliferating cells may express specific histone variants (e.g., H3.3) at elevated levels. Tissue complexity can also influence the choice of homogenization and purification steps. As a rule of thumb, 30 mg of tissue or approximately 4×10⁶ cultured cells typically yields sufficient material for MS analysis.
Step-by-Step Methods for Histone Extraction
Acid Extraction of Histones from Cultured Cells and Tissues
Acid extraction is the most widely used method for histone isolation. It exploits the solubility of histones under acidic conditions, typically using 0.2 M HCl. Acidic extraction preserves most PTMs and results in high-purity core histone fractions.
Nuclei Isolation
Effective nuclei isolation is essential to reduce cytoplasmic contamination. Cells are lysed under hypotonic conditions with non-ionic detergents, followed by centrifugation to pellet nuclei. Proper nuclear integrity ensures minimal loss of histones during extraction.
Protocol for HCl-Based Acid Extraction
- Collect and wash cells or tissue in PBS.
- Lyse in hypotonic buffer with protease inhibitors.
- Pellet nuclei by centrifugation.
- Resuspend in 0.2 M HCl and incubate for 1-2 hours at 4°C.
- Centrifuge to remove insoluble debris.
- Precipitate histones using TCA or acetone, wash, and dry.
This method yields highly enriched core histones, suitable for downstream derivatization and MS profiling.
Alternative Extraction Methods: Salt, Detergent, and Urea Protocols
- While acid extraction remains standard, alternative methods exist:
- High-salt buffers (e.g., 2 M NaCl) extract loosely bound histones.
- Urea-based protocols denature and solubilize histones from chromatin.
- Detergent-based buffers help with membrane solubilization but may disrupt PTMs.
Such methods are context-dependent and often serve as complements rather than replacements for acid extraction.
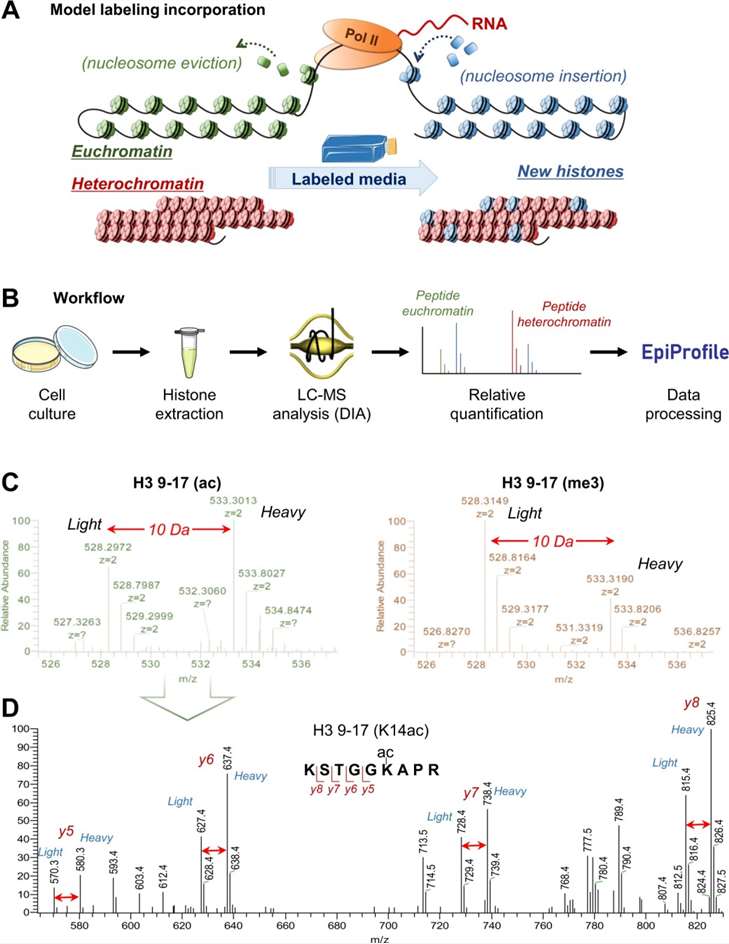
Chemical Derivatization and Sample Preparation for MS
Why Derivatize Histones Before Mass Spectrometry?
Histones are rich in lysine and arginine residues, which result in extremely short peptides upon trypsin digestion. Derivatization chemically modifies free amines, restricting cleavage to arginine residues. This step produces peptides of optimal length and improves chromatographic behavior.
Choosing the Right Reagent: Propionic vs. d₆-Acetic Anhydride
- Propionic anhydride: common and effective for acetylation and methylation studies. However, it introduces mass shifts indistinguishable from endogenous propionylation.
- d₆-Acetic anhydride: preferred when studying acylations. It avoids mass overlap and permits the quantification of endogenous propionyl or butyryl modifications.
Step-by-Step Derivatization and Trypsin Digestion Protocol
- Resuspend histones in ammonium bicarbonate buffer.
- Add propionic or d₆-acetic anhydride and incubate.
- Perform trypsin digestion overnight.
- Repeat derivatization to block newly formed N-termini.
- Clean up using C18 desalting or StageTips.
Enhancing Peptide Retention
Small histone peptides often elute early during reversed-phase liquid chromatography. Modification with The Role of Phenylisocyanate (PIC) increases peptide hydrophobicity, improving retention and separation. This step is especially useful for quantifying low-abundance peptides.
Select Service
Histone Enrichment Techniques for Downstream Applications
Affinity-Based Enrichment of Specific Histone PTMs
Affinity-based techniques allow selective enrichment of histones carrying specific PTMs. These methods often utilize immobilized antibodies, PTM-binding domains, or synthetic ligands that recognize particular chemical groups. They enhance sensitivity and facilitate the study of low-abundance modifications.
Histone peptides or intact nucleosomes can be enriched using modified peptide pull-down assays or chromatin immunoprecipitation-like approaches. These strategies are essential for mapping reader interactions or identifying PTM combinations.
Antibody-Based Pull-Down: Strengths and Limitations
Immunoprecipitation remains widely used for enriching histone PTMs. However, antibody quality is a major constraint. Many commercial antibodies lack specificity or exhibit cross-reactivity. Moreover, antibody-based enrichment is typically semi-quantitative and does not allow multiplex analysis.
Therefore, antibody-based methods are better suited for validation rather than discovery workflows. Proper validation through dot blotting or peptide competition assays is required.
Enrichment Using Chemical Tags or Biotinylated Peptides
Chemical tagging techniques introduce biotin or reactive groups onto PTMs or modified amino acids. These tags enable streptavidin-mediated capture and isolation. Examples include azide-alkyne click chemistry for acylations and thiol-reactive probes for cysteine-containing modifications.
Such approaches allow efficient enrichment and can be used in conjunction with MS or western blotting. They offer greater control over specificity and enable parallel screening of multiple modifications.
Offline Fractionation of Histone Variants
Histone variants differ in sequence, expression, and function. Separation of variants such as H3.1 and H3.3 requires offline fractionation. This can be achieved through ion exchange chromatography, size exclusion, or isoelectric focusing.
Variant-specific separation enhances the detection of PTMs that are uniquely associated with particular histone forms. It is especially important for studies involving developmental stages or cancer subtypes where variant dynamics are critical.
Quantitative Mass Spectrometry for Histone Profiling
Bottom-Up and Top-Down Proteomics
Bottom-up proteomics involves the digestion of histones into peptides followed by MS analysis. It is the most commonly used approach due to its high sensitivity and compatibility with labeling strategies. However, it cannot resolve combinatorial PTMs on the same histone molecule.
Top-down proteomics analyzes intact proteins without the digestion. It enables detection of all modifications present in a single protein species. Despite its analytical power, it requires high-resolution instruments and advanced separation techniques.
The choice between the two depends on the research objective. Bottom-up is ideal for large-scale screening. Top-down is suited for studies requiring intact PTM mapping.
Labeling Strategies: SILAC, TMT, iTRAQ, and Label-Free Methods
- SILAC: Incorporates heavy isotopes into proteins during cell growth. Enables accurate relative quantification.
- TMT/iTRAQ: Chemical tags introduced post-digestion. Allow multiplexing of multiple samples in one MS run.
- Label-Free: Quantifies peptides based on spectral intensity or ion counts. Requires rigorous normalization and batch consistency.
MS Instrumentation
- DDA: Selects the most intense precursor ions for fragmentation. Provides high specificity but can miss low-abundance peptides.
- DIA: Fragments all ions within set windows. Offers comprehensive coverage and reproducibility.
DIA is gaining prominence in the study of histone PTMs owing to its capacity to identify infrequent modifications and minimize the occurrence of missing values across replicate analyses.
Data Processing Tools: EpiProfile 2.0, Skyline, and R Workflows
- EpiProfile 2.0: Automated software tailored to histone peptide analysis. Provides accurate peak identification for well-separated, high-quality MS data.
- Skyline: Enables manual peak inspection. Offers flexibility for complex spectra but requires user expertise.
- R-based Workflows: Custom scripts facilitate normalization, statistical filtering, and visualization. Suitable for batch processing and reproducibility checks.
Select Service
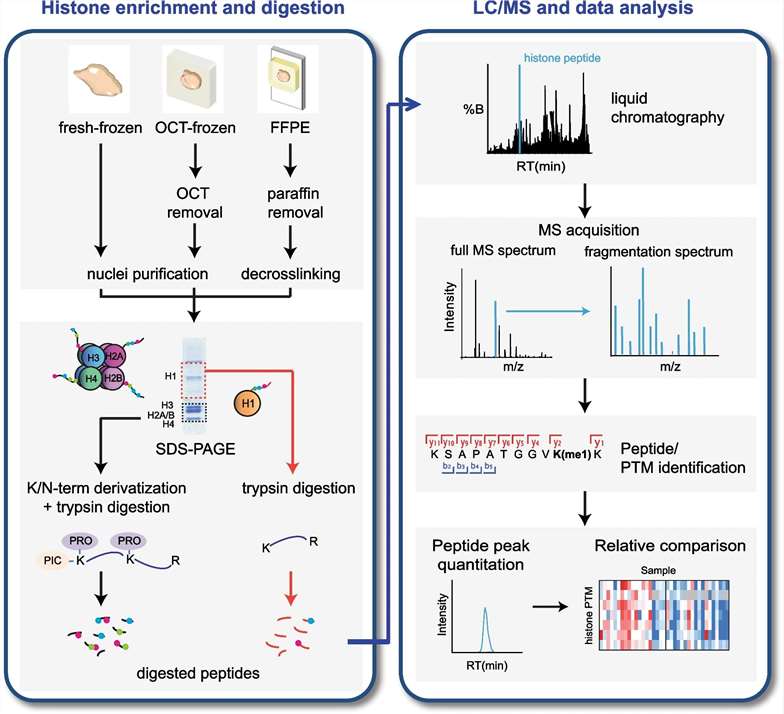 Figure 1. Schematic bottom-up workflow for MS-based analysis of histone PTMs and variants (Robusti G, et al., 2022).
Figure 1. Schematic bottom-up workflow for MS-based analysis of histone PTMs and variants (Robusti G, et al., 2022).
Troubleshooting Low-Yield or Contaminated Histone Samples
Diagnosing Low Purity
Histone extracts with low purity often contain cytoplasmic or nuclear debris. Poor nuclei isolation or over-digestion during sample preparation may contribute. Indicators of low purity include visible protein smearing, excessive background in MS spectra, and inconsistent PTM signals.
Dealing with Poor MS Signal or Co-Eluting Contaminants
Low signal intensity may result from protein degradation, incomplete derivatization, or inefficient digestion. Co-eluting contaminants can obscure target peptides and impair quantification. Ensuring rigorous cleanup steps and consistent sample handling is critical.
Best Practices for Sample Storage and Handling
Histones should be stored at −80°C for long-term preservation. Repeated freeze-thaw cycles must be avoided. Sample derivatization and digestion should be performed on ice or under cold conditions. The use of protease and deacetylase inhibitors is mandatory during extraction and handling.
Case Study
Enrichment of histones from patient samples for mass spectrometry-based analysis of post-translational modifications
Journal: Clinical Epigenetics
Published: 2019
DOI: 10.1016/j.ymeth.2019.10.001
Background
Aberrations in histone PTMs are implicated in diverse pathologies, including cancer. Profiling PTMs in clinical specimens—such as formalin-fixed paraffin-embedded (FFPE) tissues or primary cells—is technically challenging. Traditional antibody-based assays (e.g., Western blotting, ChIP) are targeted and semi-quantitative. In contrast, mass spectrometry (MS) offers broader, unbiased, and quantitative insight into histone PTMs.
Purpose
The study aims to develop and validate protocols for extracting and enriching histones from multiple types of patient-derived specimens—FFPE, frozen tissues, and primary cells—to enable robust MS-based PTM profiling suitable for clinical and biomarker research.
Methods
The authors describe three extraction workflows tailored to different sample types:
- FFPE tissues: Employ deparaffinization and protein retrieval, followed by histone enrichment.
- Fresh or frozen tissues: Implement cryogenic homogenization, nuclear isolation, and extraction buffers optimized for histone solubilization.
- Primary cells: Use detergent-mediated nuclear isolation followed by acid extraction (e.g., 0.2 M HCl).
Each workflow concludes with acetone or TCA precipitation. Histone-enriched pellets are then digested (trypsin) and analyzed by LC-MS/MS. The authors report expected protein recovery and PTM detection rates for each sample category.
Results
The optimized protocols consistently yielded histone preparations amenable to MS analysis across various clinical specimen types.
PTM coverage, including acetylation and methylation marks, was comparable across FFPE, frozen tissue, and primary cell samples.
The workflows demonstrated reproducibility, showing that clinically relevant archived samples can yield high-quality histone PTM profiles.
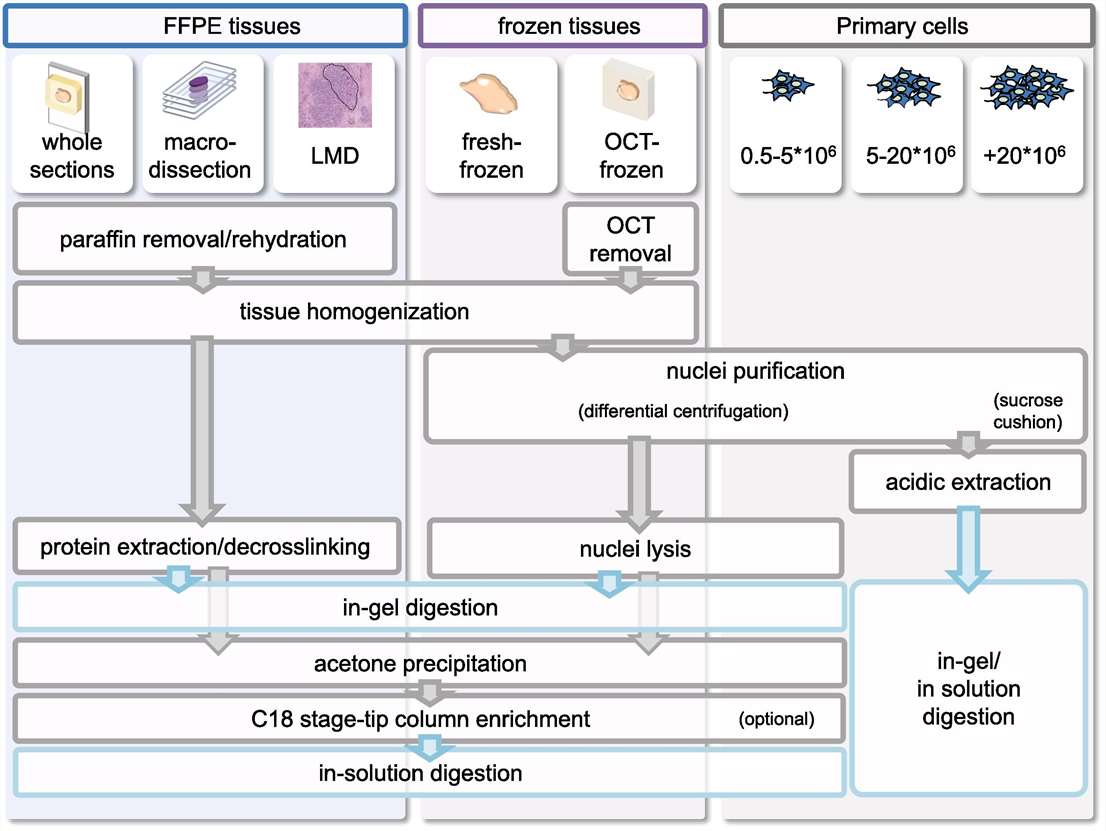 Figure 2. Overview of protocols for the isolation and enrichment of histones from primary samples prior to MS analysis.
Figure 2. Overview of protocols for the isolation and enrichment of histones from primary samples prior to MS analysis.
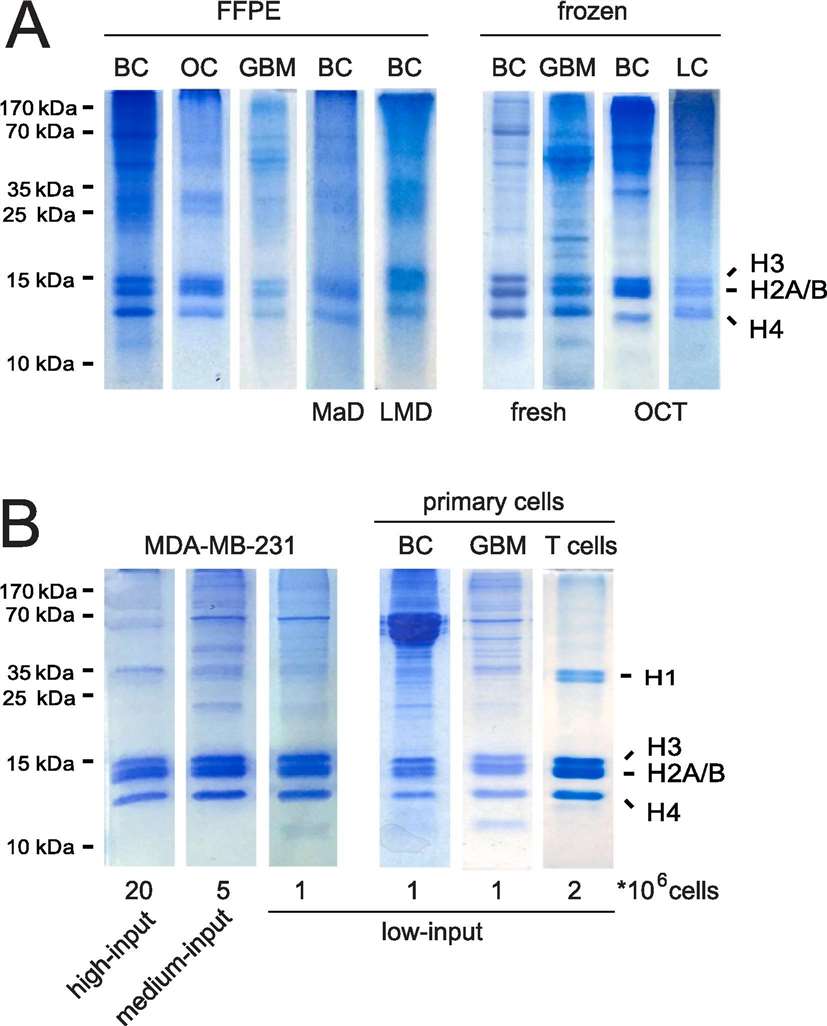 Figure 3. Histones isolated from primary samples through different protocols.
Figure 3. Histones isolated from primary samples through different protocols.
Conclusions
The authors successfully established MS-ready protocols for histone enrichment from clinically relevant biospecimens.
These methods overcome obstacles associated with sample preservation (e.g., formalin fixation) and limited input material.
References
- Huang H, et al. Quantitative proteomic analysis of histone modifications. Chemical reviews, 2015, 115(6): 2376-2418. DOI: 10.1021/cr500491u
- Robusti G, et al. Investigating pathological epigenetic aberrations by epi-proteomics. Clinical Epigenetics, 2022, 14(1): 145. DOI: 10.1186/s13148-022-01371-y
- Noberini R, et al. Enrichment of histones from patient samples for mass spectrometry-based analysis of post-translational modifications. Methods, 2020, 184: 19-28. DOI: 10.1016/j.ymeth.2019.10.001

Related Articals
Our products and services are for research use only.
