
What Are Histone Post-Translational Modifications (PTMs)?
Histone PTMs refer to covalent chemical modifications that occur on histone proteins after translation. These modifications include acetylation, methylation, phosphorylation, ubiquitination, and many others. They predominantly affect lysine, arginine, serine, threonine, and tyrosine residues. These chemical changes do not alter the DNA sequence. However, they play critical roles in regulating chromatin architecture and genomic function.
Histone PTMs occur mainly on the N-terminal tails of histone proteins. These tails extend outward from the nucleosome core. They serve as platforms for the recruitment of protein complexes. Through this mechanism, histone PTMs exert influence over gene transcription, DNA replication, repair, and recombination. The diversity and reversibility of these modifications contribute to the plasticity of the chromatin landscape.
Why Histone PTMs Quantification Matters
The mere presence of a histone PTM is insufficient to explain its functional relevance. Quantitative analysis is necessary to determine dynamic changes in modification patterns. Such changes may reflect cell state, environmental stimuli, or disease progression.
Traditional techniques, such as Western blotting and immunofluorescence, offer only semi-quantitative outputs. These methods are also limited by antibody specificity. Mass spectrometry (MS), in contrast, provides an unbiased and highly sensitive platform. It enables precise quantification across a wide array of PTMs in a single analysis. Quantitative analysis supports hypothesis-driven research. It allows investigators to distinguish between basal and altered epigenetic states. Furthermore, it provides data essential for systems biology and biomarker discovery.
Select Service
Biological Significance of Histone Modification Dynamics
Histone Code and Chromatin Structure Regulation
Histone PTMs act in combinations, forming a regulatory language termed the "histone code." This code influences chromatin accessibility and higher-order organization. Specific combinations of PTMs may promote open euchromatin or condensed heterochromatin. In this way, PTMs function as molecular switches to regulate DNA availability.
Metabolic Control of PTM "Writers" and "Erasers"
Histone-modifying enzymes require metabolites as cofactors. Acetyltransferases utilize acetyl-CoA, while sirtuins depend on NAD⁺. Methyltransferases use S-adenosylmethionine as a methyl donor. Therefore, fluctuations in metabolite levels can directly affect histone PTM patterns. This establishes a functional link between metabolic state and chromatin structure.
PTMs as Disease-Associated Epigenetic Signatures
Aberrant histone PTM profiles have been associated with various pathologies. These include cancers, metabolic disorders, neurodegeneration, and autoimmune diseases. Identifying disease-specific PTM signatures may facilitate early diagnosis and therapeutic stratification. In cancer, for instance, hyperacetylation or loss of repressive methylation may drive oncogenic transcription programs.
Bottom-Up LC-MS/MS Strategies for Histone PTM Profiling
Bottom-up proteomics is the most commonly employed approach for extensive, site-specific examination of histone PTMs. Its sensitivity, reproducibility, and scalability make it the cornerstone of the majority of workflows for quantifying histone PTMs.
Histone Extraction and Purity Requirements
Accurate PTM analysis begins with high-purity histone extraction. Histones are tightly associated with DNA and nuclear proteins, necessitating an effective isolation protocol. The most commonly employed method is acid extraction using H₂SO₄ or N HCl. This treatment selectively solubilizes histones while precipitating most non-histone proteins.
Purity is critical. Impurities such as transcription factors or non-histone chromatin components can interfere with downstream labeling and MS detection. Therefore, histone preparations should exceed 80% purity, as confirmed by SDS-PAGE or reversed-phase HPLC.
Chemical Derivatization
Histones contain a high density of lysine and arginine residues, which are the natural cleavage sites for the protease trypsin. Without chemical modification, trypsin digestion generates excessively short peptides that are unsuitable for MS analysis due to poor retention and ionization.
To overcome this issue, chemical derivatization is used to block lysine residues, thereby restricting trypsin cleavage exclusively to arginine sites. This results in peptides of optimal length and improved chromatographic behavior.
Propionic Anhydride or d₆-Acetic Anhydride Labeling
Propionic anhydride reacts with free amino groups on unmodified lysines and N-termini, converting them into propionyl groups. This approach ensures standardized cleavage patterns and enhances peptide hydrophobicity for improved retention in reversed-phase liquid chromatography (RPLC).
However, a critical drawback of propionic anhydride is that it introduces a mass shift, which is identical to that of endogenous lysine propionylation. This leads to ambiguity when detecting natural propionyl modifications.
To address these limitations, d₆-acetic anhydride is used as an alternative. It introduces a heavy acetyl group that is easily distinguishable from natural PTMs. This reagent allows for accurate quantification of acetylation, propionylation, and butyrylation, as it avoids overlapping mass shifts.
Phenyl isocyanate (PIC) Capping for Peak Resolution
Following digestion and derivatization, the resulting peptides are chemically modified at their N-termini with PIC. This step introduces a hydrophobic phenylcarbamate moiety that enhances peptide separation and sharpens peak resolution in RPLC.
The PIC modification:
- Increases peptide retention time
- Reduces co-elution of isobaric species
- Improves signal-to-noise ratio in MS detection
This step is especially valuable when analyzing short peptides, which often elute rapidly and exhibit poor separation without hydrophobic tagging.
Trypsin Digestion and Peptide Cleanup
Proteolysis is performed using sequence-grade trypsin, which cleaves C-terminal to arginine residues under the modified conditions. Following digestion, peptide cleanup is essential to remove salts, derivatization reagents, and small molecule contaminants. Solid-phase extraction (SPE) using C18 desalting tips or cartridges is routinely employed. This step concentrates the peptides and improves ionization efficiency during MS analysis.
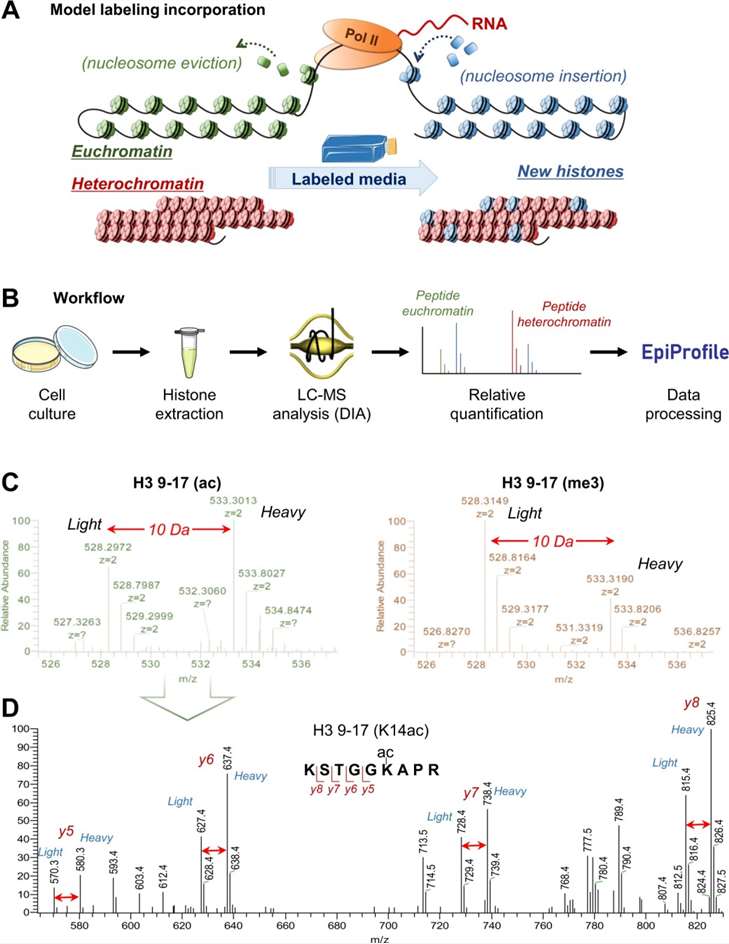 Figure 1. Overview of the workflow for bottom-up MS/MS histone analysis (Sidoli S, et al., 2019).
Figure 1. Overview of the workflow for bottom-up MS/MS histone analysis (Sidoli S, et al., 2019).
Top-Down and Middle-Down MS for Combinatorial PTM Mapping
Conventional bottom-up MS strategies fragment histones into short peptides, which often masks this combinatorial information by separating modifications that co-occur in vivo. In contrast, Top-down and middle-down proteomics enables the direct analysis of intact histone proteins or large polypeptides. These approaches preserve the full PTM landscape of individual histone isoforms and allow for a deeper understanding of how PTMs cooperate in regulating chromatin function.
ECD/ETD Fragmentation for Labile Modifications
Top-down and middle-down workflows require advanced fragmentation techniques capable of preserving labile PTMs, such as phosphorylation, O-GlcNAcylation, and crotonylation. Two electron-based dissociation methods are commonly employed:
- Electron Capture Dissociation (ECD) introduces low-energy electrons to multiply charged precursor ions, producing c- and z-type fragment ions without disrupting labile side chains.
- Electron Transfer Dissociation (ETD) achieves similar outcomes by transferring electrons from anionic reagents to the precursor ion.
Both techniques enable site-specific localization of PTMs and are particularly suited for the analysis of multiply modified histones. Importantly, ECD and ETD generate rich fragmentation data that can distinguish between isobaric modifications and resolve closely spaced PTMs on the same peptide or protein backbone.
Advantages of Intact-Protein Analysis
Top-down proteomics involves the direct introduction and analysis of intact histone proteins (typically 10-20 kDa) without enzymatic digestion. This technique provides:
- Complete sequence coverage, capturing all PTMs on a single molecule.
- Direct mapping of PTM crosstalk, including co-occurrence, mutual exclusivity, and modification order.
- Precise proteoform-level resolution, allowing for the identification of distinct histone variants or splice forms.
Such resolution is essential for interpreting complex PTM patterns, especially for histone H3 and H4, which exhibit extensive acetylation and methylation in overlapping positions.
Middle-Down MS
Middle-down proteomics offers a compromise between the full coverage of top-down and the scalability of bottom-up approaches. In this strategy:
- Histones are cleaved using proteases such as GluC, AspN, or ArgC, which generate large peptides (3-10 kDa).
- These peptides retain multiple modification sites while reducing spectral complexity and improving chromatographic performance.
- Middle-down MS allows for better coverage of PTMs on long N-terminal tails, such as the H3(1-50) peptide, which carries the majority of functionally relevant PTMs.
Recent studies have demonstrated that middle-down MS can successfully capture combinatorial acetylation and methylation states on histone H3 and H4 tails, revealing novel regulatory signatures linked to transcriptional activity or repression.
Select Service
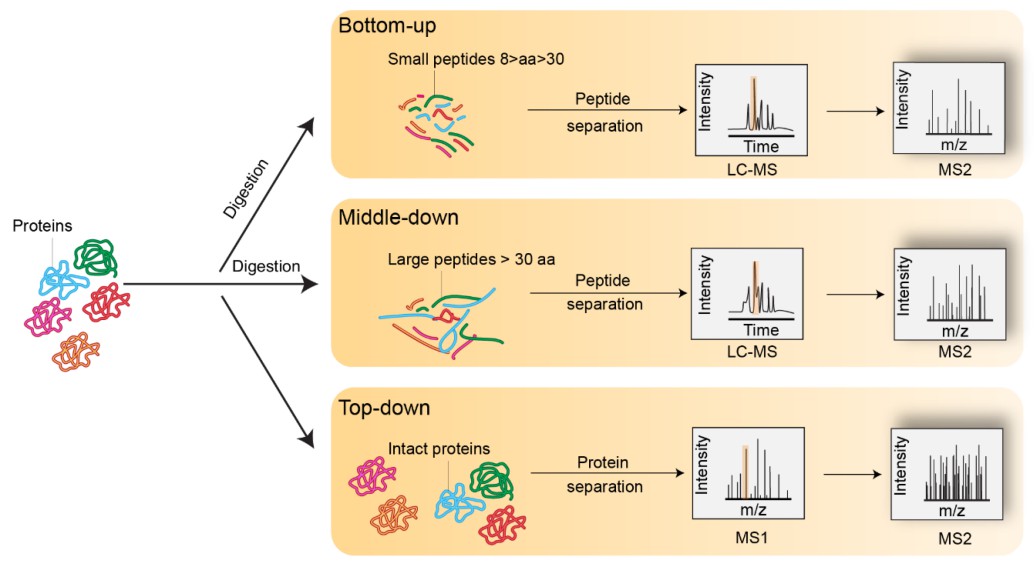 Figure 2. Representation of bottom-up, middle-down, and top-down MS experiments (El Kennani S, et al., 2018).
Figure 2. Representation of bottom-up, middle-down, and top-down MS experiments (El Kennani S, et al., 2018).
Comparison of Histone MS Strategies
| Strategy | Bottom-up | Middle-down | Top-down |
|---|---|---|---|
| Analyte Size | Small peptides | Large peptides | Full-length proteins |
| PTM Context | Lost | Partial retained | Fully retained |
| Fragmentation | CID/HCD | ETD/ECD preferred | ECD/ETD required |
| Advantages | High sensitivity, widely used | Preserves partial PTM context, good sequence coverage | Complete PTM map, no digestion bias |
| Limitations | PTM combinations lost, derivatization required | Sample complexity, moderate instrument demand | Low sensitivity, high complexity, expensive tools |
Labeling Approaches for Quantitative Comparison
In Vivo Isotope Labeling
SILAC incorporates heavy lysine and arginine. It yields "light" and "heavy" cell populations. It enables accurate pairwise quantification. It minimizes technical variation.
In Vitro Isobaric Tagging
TMT/iTRAQ reagents react with peptide N-termini and lysine side chains. They produce isobaric precursors. Reporter ions cleave in MS2 to reveal sample origin. They permit multiplexing of up to 16 samples.
Dimethyl and DiLeu Chemical Tags
Dimethylation uses formaldehyde and cyanoborohydride to add light or heavy methyl groups. DiLeu tags employ leucine derivatives with differential isotopic composition. Both approaches are cost-effective alternatives for 4- or 8-plex quantification.
Label-Free Quantification: MS1 vs. MS2 Workflows
Label-free methods derive abundance from precursor MS1 peak areas or MS2 spectral counts. Data-independent acquisition (DIA) offers comprehensive MS2 coverage. It reduces missing values compared to data-dependent acquisition (DDA).
Select Service
Data Acquisition: DDA versus DIA for Histone Peptides
Data-Dependent Acquisition (DDA) Fundamentals
DDA selects the top N precursor ions for MS2 fragmentation. It favors high-abundance peptides. It can miss low-level PTMs in complex samples.
Data-Independent Acquisition (SWATH/DIA) Benefits
DIA fragments all precursors in predefined windows. SWATH-MS records MS2 for every detectable precursor. It achieves deep coverage of low-abundance PTMs and high reproducibility.
Minimizing Batch Effects and Retention-Time Drift
Randomize sample order within batches. Include a pooled quality-control sample every 5-10 runs. Monitor retention times using standard peptides to correct drift.
Bioinformatics and Software Tools for Histone PTM Analysis
Automated Peak Picking
EpiProfile 2.0 identifies histone peptides and associated PTMs. It extracts MS1 peak areas automatically. It accelerates analysis but may omit medium-quality peaks.
Manual Integration with Skyline for Site-Specific Quantification
Skyline allows visual inspection of MS1 and MS2 peaks. It supports custom transition lists. It delivers precise quantification for peptides with multiple lysines.
Non-Restricted Sequence Alignment for Novel PTM Discovery
Algorithms such as InsPecT and PTMap scan MS2 spectra for unknown mass shifts. They enable discovery of the novel PTM without prior specification.
R-Based Pipelines: Quality Control, Normalization, and Statistics
Custom R scripts perform data checking, normalization (e.g., total ion current), and significance testing. They apply filters for benchmark peptides. They control the false-discovery rate and generate publication-ready reports.
Select Service
Applications in Disease and Development
Quantifying PTM Dynamics in Cancer Cell Lines
Aberrant histone PTM landscapes are a hallmark of cancer. Dysregulation of histone acetylation, methylation, and ubiquitination has been documented across multiple tumor types. Quantitative mass spectrometry allows for the comparative profiling of histone PTMs in cancer cell lines under various treatments or genetic perturbations.
Biomarker Discovery in Metabolic Disorders
Histone acylations—such as crotonylation, propionylation, and succinylation—are emerging as sensitive indicators of metabolic flux. These PTMs are derived from metabolic intermediates, including crotonyl-CoA, propionyl-CoA, and succinyl-CoA.
Epigenetic Profiling in Stem Cell Differentiation
Stem cell fate decisions are governed by dynamic remodeling of histone PTM. During differentiation, pluripotent cells resolve bivalent chromatin domains—regions marked by both H3K4me3 (activation) and H3K27me3 (repression)—into univalent states. This epigenetic resolution is essential for lineage specification.
Case
Case 1: Quantitative proteomic analysis of histone modifications in decitabine sensitive and resistant leukemia cell lines
Citation: Zhang C, et al. Clinical proteomics, 2016, 13: 1-11. DOI: 10.1186/s12014-016-9115-z
The authors developed a robust bottom-up LC-MS/MS workflow optimized for leukemia cell lines. They aimed to quantitatively profile histone marks in decitabine-sensitive and -resistant cells. They sought to identify PTM patterns that could underlie drug response or resistance.
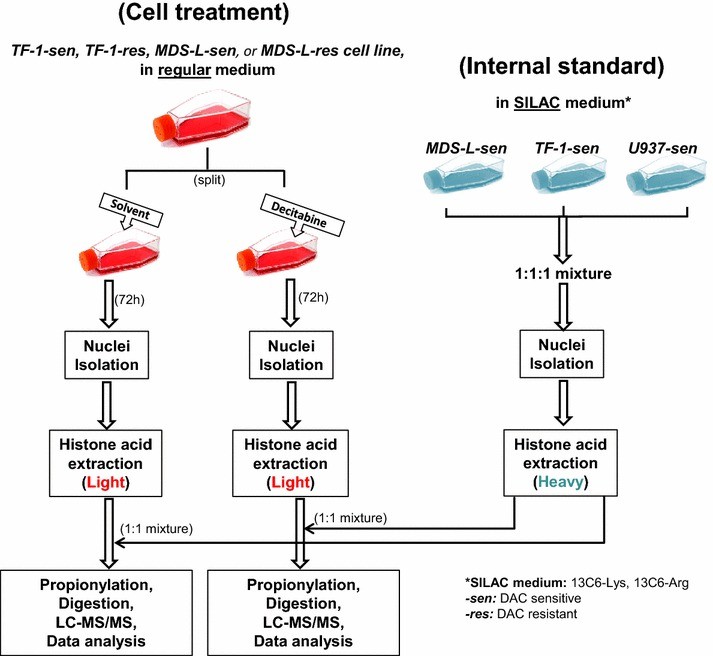 Figure 3. Experimental design for analysis of histone modifications in decitabine sensitive and resistant leukemia cell lines.
Figure 3. Experimental design for analysis of histone modifications in decitabine sensitive and resistant leukemia cell lines.
Case 2: Top-down and Middle-down Protein Analysis Reveals that Intact and Clipped Human Histones Differ in Post-translational Modification Patterns
Citation: Tvardovskiy A, et al. Molecular & Cellular Proteomics, 2015, 14(12): 3142-3153. DOI: 10.1074/mcp.M115.048975
This study provided the first direct evidence that histone clipping is associated with distinct PTM patterns in human liver cells. The findings suggest that proteolytic histone processing contributes to chromatin remodeling and epigenetic regulation in spatially structured tissue environments. The combined top-down and middle-down MS approach enables comprehensive proteoform characterization.
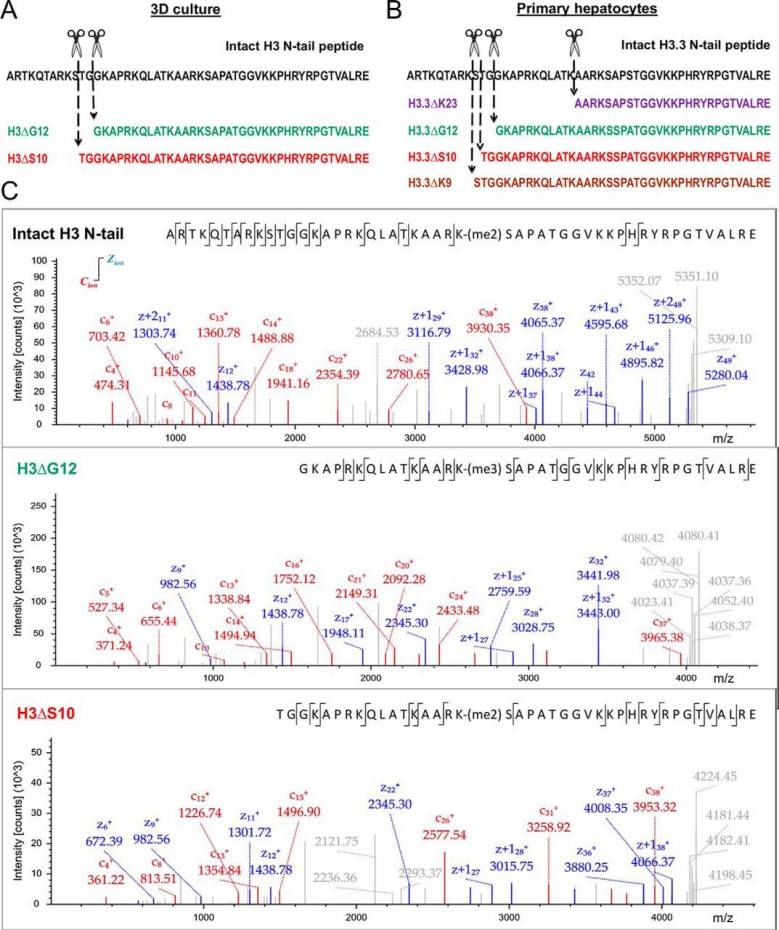 Figure 4. Determination of H3 and H2B clipping sites by middle-down MS.
Figure 4. Determination of H3 and H2B clipping sites by middle-down MS.
References
- Sidoli S, et al. A mass spectrometry-based assay using metabolic labeling to rapidly monitor chromatin accessibility of modified histone proteins. Scientific reports, 2019, 9(1): 13613. DOI: 10.1038/s41598-019-49894-4
- Sidoli S, et al. Complete workflow for analysis of histone post-translational modifications using bottom-up mass spectrometry: from histone extraction to data analysis. Journal of visualized experiments: JoVE, 2016 (111): 54112. DOI: 10.3791/54112
- El Kennani S, et al. Proteomic analysis of histone variants and their PTMs: strategies and pitfalls. Proteomes, 2018, 6(3): 29. DOI: 10.3390/proteomes6030029
.jpg)

Our products and services are for research use only.
