
Since the last century, our understanding of post-translational modifications (PTMs) in proteins has significantly deepened. Over the past decade, further insight has been gained through the discovery of novel protein PTMs, the novel development of whole-genome maps, and the inventive strategies for localized and temporal control of PTM deposition or removal. In this digital age, with the widespread availability of vast amounts of data from various biological systems, the functional roles of PTMs are gradually being revealed in key processes such as transcription, recombination, replication, DNA repair, and genomic structural regulation.
With a broad array of types, post-translational modifications exhibit unique features specific to various proteins. Methylation is the most common form of modification that influences protein structure and interactions with other proteins, implicated in virtually all biological processes. Phosphorylation, on the other hand, can alter protein conformation, activating or inhibiting catalytic activity. Recently, novel areas of research and emerging trends are becoming more noticeable involving other forms of modifications. In this paper, we will be focusing, by way of example, on ubiquitination among other PTMs, elucidating on their functionalities, current research focuses, and selective investigative strategies.
Select Service
Acetylation
Acetylation impacts protein conformation and affinity with other proteins, participating in the regulation of transcription and metabolism.
Nε Acetylation, colloquially termed "acetylation", is a pervasive and reversible PTM, orchestrated by two classes of enzymes: Lysine acetyltransferases (KATs) and Lysine deacetylases (KDACs). KATs primarily catalyze the transfer of acetyl groups onto Lys residues, while KDACs facilitate the removal of acetyl groups. The KDACs are subdivided into two subgroups: the Zn2+ dependent histone deacetylases (HDACs) and the NAD+-dependent sirtuins. More than 40 different Lys residues in the four canonical core histones H2A, H2B, H3, and H4 can be acetylated. Additionally, proteomic studies have revealed acetylation phenomena in thousands of non-histone proteins in the cell, affecting processes such as metabolism, RNA processing, translation, protein folding, chromatin organization, protein degradation, and cytoskeleton organization. Recent studies also view acetylation as a means to identify candidate disease biomarkers and have found that histone acetylation can mediate cellular memory. This capacity for acetylation permits the coupling of cell state with transcriptional output and cell fate decisions.
PTMs like phosphorylation, ubiquitination, and Acetylation play significant roles in autophagy regulation, with acetylation being a pivotal regulatory mechanism of autophagy. Acetylation influences autophagy initiation and autophagosome formation by targeted regulation of core components like ULK1 complex, BECN1-PIK3C3 complex, and LC3 lipidation system. Further, recent research suggests that acetylation occurs on key proteins involved in autophagic assembly and autophagosome-lysosome fusion, such as SQSTM1/p62 and STX17. Additionally, acetylation also regulates autophagy at the transcriptional level by targeting histones and the transcription factor TFEB.
Select Service
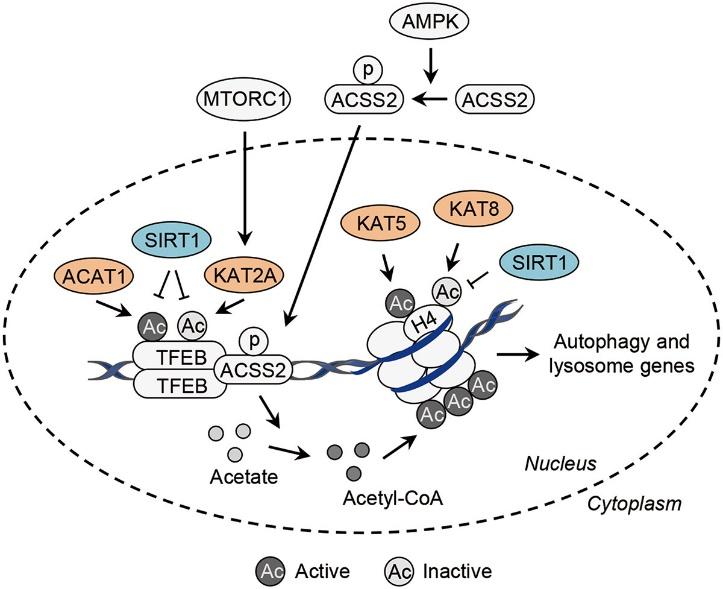 Transcriptional control of autophagy-related genes by acetylation.
Transcriptional control of autophagy-related genes by acetylation.
Ubiquitination: An Essential Process in Protein Degradation
Ubiquitination, the covalent linkage of ubiquitin to proteins, is a prevalent post-translational modification (PTM) with significant implications for the stability, subcellular positioning, functionality, and complex formation of substrate proteins. The ubiquitination process is catalyzed by a collection of enzymatic reactions involving ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin-ligating enzymes (E3). In the final step, an isopeptide bond forms between the carboxyl group of the terminal glycine residue on ubiquitin and the epsilon amino group of a lysine residue in the substrate protein.
The ubiquitin superfamily is a tremendous set of proteins, also involving autophagy-related (ATG) 8, ATG12, neural precursor cell expressed developmentally down-regulated protein 8 (NEDD8), small ubiquitin-related modifier (SUMO), human leukocyte antigen F adjacent transcript 10 (FAT10), among others. This superfamily also includes the smaller subfamilies of interferon-stimulated gene 15 kDa (ISG15) and ubiquitin-fold modifier 1 (UFM1). Like ubiquitin, these family proteins also establish covalent links with their substrates through analogous ubiquitin system reactions.
Recent studies have directed attention to the formation of ester bonds between ubiquitin's serine or threonine residues (rather than the terminal glycine) and phospholipids, while the exact connection site remains to be identified. A recent investigation has revealed that mammalian cells' host E3 ligase, the RING finger (RNF) 213, ubiquitinates Salmonella lipopolysaccharide, facilitating bacterial removal via autophagy. In this case, ubiquitin may form an ester bond with lipopolysaccharides instead of a typical peptide bond, but the precise linkage mode remains undetermined.
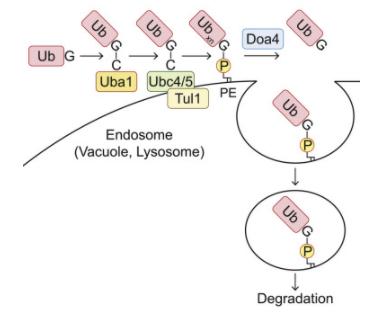
Within the ubiquitin family, the ATG8 subfamily uniquely features a prominent association with phospholipids. Existing research has unveiled that ubiquitin forms bonds with phospholipids, notably phosphatidylethanolamine (PE), in both yeast and mammalian cells. To investigate whether ubiquitin interacts with organelle membrane phospholipids, it is feasible to purify ubiquitin species possessing hydrophobic traits from samples. For instance, solubilizing the cell membrane from cells expressing 3×FLAG-ubiquitin in a buffer containing 1% TritonX-114, followed by heating to the cloud point to achieve phase partitioning. Ubiquitin can then be immunoprecipitated from the TritonX-114 fraction using an anti-FLAG antibody. In addition to a substantial amount of polyubiquitin species, a band with a higher molecular weight (Ub*) can also be detected in the hydrophobic sector of TritonX-114. After treatment with phospholipase D (PLD), which hydrolyzes the diester bond of glycerophospholipids, this high molecular weight entity shifts downwards, thereby confirming the association of ubiquitin with phospholipids.
To detect ubiquitinated phospholipids at endogenous levels within cells, hydrophobic ubiquitin species can be recovered from yeast membranes through affinity purification using ubiquitin-related (UBA) domain of ubiquilin 1 (UQ1). Diand triubiquitin conjugates, which can be cleaved by PLD, can be detected in the TritonX-114 detergent phase. The absence of ubiquitin monomers in the TritonX-114 phase could be attributed to the attributes of UQ1UBA, as it tends to capture oligo- and polyubiquitin rather than monoubiquitin. Moreover, the multiphospholipidation status of endogenous phospholipids can be studied via lipid extraction assays.
To determine the types of phospholipids that bind to ubiquitin in the sample, phosphatidylserine (PS) and PE can be considered, as they are potential ubiquitination targets, having an amino group capable of forming an amide bond with the C-terminal glycine of ubiquitin and known to bind with ATG8 family proteins. Subsequently, gel-internal saponification (acyl chain cleavage), leucine aminopeptidase digestion, and tandem mass spectrometry (MS/MS) analysis can be performed on Ub*.
To ascertain the location of Ub-PE, cell membranes can be isolated via membrane flotation in discontinuous iodixanol gradients, followed by TritonX-114 phase partitioning. When further identifying the enzyme catalyzing PE ubiquitination, the auxin-induced degron system can be adopted to deplete ubiquitin-activating enzyme (E1) Uba1, to inhibit PE ubiquitination. To confirm that the identified enzyme catalyzes the sufficient transfer of ubiquitin to PE, the process can be reconstituted in vitro using liposomes composed of 40 mol% PE or PS and 50 mol% phosphatidylcholine (PC).
For a more thorough investigation of whether the hydrophobic ubiquitin species detected in the cell indeed contain Ub-PE, Ub-PE reconstituted in vitro can serve as a molecular standard. By using MS/MS spectra of C-terminal peptides with or without GPE modifications derived from in vitro reconstituted Ub-PE (post-saponification and leucine aminopeptidase treatment), peptide quantification based on mass spectrometry can be established through a combination of selective reaction monitoring (SRM) method and stable isotope labeling. Subsequently, further investigations can then explore whether other ubiquitin-like proteins are also binding to phospholipids, thereby expanding on the existing findings.
Select Service
Sumoylation: Subcellular Localization and Protein Interactions
Sumoylation, a modification involving small ubiquitin-related modifiers (SUMOs), is an emerging form of ubiquitin-like molecular modification. Like ubiquitination, it is integral to crucial post-translational modifications (PTMs). Sumoylation events are catalyzed by a minor modification enzyme, dynamically regulating a multitude of target proteins. SUMOs can attach to target proteins either as individual monomers or in various types of polymeric forms. Non-covalent receptors recognize Sumoylated proteins via SUMO interaction motifs.
Sumoylation acts upon functionally partnered proteins to control core nuclear processes, encompassing gene expression, the DNA damage response, RNA processing, the progress of the cell cycle, and protein stability. Recent research advances have deepened our understanding of the cellular and pathophysiological roles of Sumoylation, extending its function to the regulation of immunity, pluripotency, and nucleolar assembly in response to oxidative stress, partly realized through the recently characterized liquid-liquid phase separation mechanism.
The breakthrough in understanding the action and regulation of Sumoylation has paved new paths for disease treatment targeting Sumoylation. The first Sumoylation inhibitor is currently in clinical trials and is being explored as a potential anti-cancer drug.
Select Service
Glycosylation
The process of Glycosylation, particularly the transportation of proteins to functional regions, is complex and yet to be thoroughly investigated. The GalNAc-type O-glycosylation is of paramount importance in biology, and its aberrations are evident in truncated O-glycans in cancer. For instance, the glycoprotein MUC1 is overexpressed in most tumor tissues and tends to carry simple oligosaccharides, allowing the presentation of various tumor-associated antigens such as Tn or sTn. In contrast, tumor-induced calcification associated with O-glycosylation of fibroblast growth factor 23 presents with insufficient or low levels of O-glycans.
Significant strides have been made in deciphering the three-dimensional structures of GalNAc biomolecules, such as antibodies, lectins, mucinases, GalNAc transferases, and other glycosyltransferases. The analysis of these entities in combination with GalNAc-containing glycopeptides, primarily obtained via crystallographic or NMR analysis, contributes to understanding the key structural elements governing the recognition of glycopeptides.
Typically, the recognition of GalNAc-containing glycopeptides relies on hydrogen bonding between the GalNAc hydroxyl groups and the protein, as well as CH-π contacts, whereby the hydrophobic α-face of the sugar and the NHAc methyl group may be involved. Notably, glycopeptide binding predominantly relies on the recognition of the sugar component. However, exceptions exist, such as primary peptide recognition that utilizes sugar to enhance shape complementarity or create a limited number of interactions with anti-MUC1 antibodies.
A special attention is given to the GalNAc component, revealing degraded interactions within the same protein family, likely due to substrate flexibility. Yet, despite commonalities among families, distinguishing between them is challenging beyond common residues such as Tyr, His, or Asp responsible for hydrogen bonding. The conformational differences observed in solutions of glycopeptides carrying either GalNAc-α-1-O-Ser or GalNAc-α-1-O-Thr also manifest in the binding state.
An example illustrates this unique feature: the enzyme C1GalT1 extensively glycosylates both types of receptor substrates. This research area underscores the intricate landscape of Glycosylation, paving the way for further exploration of protein transportation and glycan functionalization.
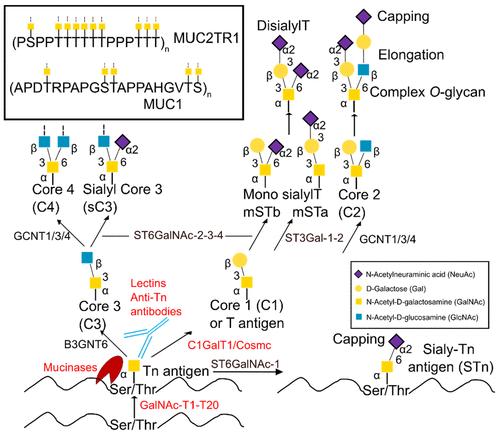 Scheme of the mucin-type O-glycosylation pathway.
Scheme of the mucin-type O-glycosylation pathway.
In the future, informed by thoughtful structure-guided studies, it will be possible to develop therapeutic vaccines and innovative diagnostic tools for early cancer detection, as well as investigate potential effective methods of cancer treatment using customized drugs targeting Tn or STn antibodies and novel inhibitors of GalNAc.
Select Service
Palmitoylation: Its influences on protein localization
Palmitoylation constitutes a post-translational lipid modification process of proteins. Through this process, a fatty acid (typically a 16-carbon palmitate ester) can be reversibly linked to cysteine residues via a thioester bond. Protein palmitoylation is catalyzed by an enzymatic group known as DHHC-palmitoyl transferases, which have an iconic DHHC motif. Numerous studies suggest that this reversible and dynamic palmitoylation plays a role in altering the protein's membrane binding or conformational state, thereby influencing subcellular protein localization and signal transduction. In the cGAS/STING signaling pathway, the palmitoylation of STING is critically important for the recruitment and activation of TBK1, contributing to the triggering of innate immune responses. However, it remains unclear as to whether cGAS undergoes palmitoylation and if this modification modulates cGAS function.
To corroborate the hypothesis of cGAS palmitoylation, the acyl-Resin Assisted Capture (acyl-RAC) assay was firstly employed to detect the Palmitoylation of cGAS. In this method, the free thiol group of cysteine in over-expressed cGAS is irreversibly blocked by N-Ethylmaleimide (NEM), revealing the hydroxylamine of the acylated cysteine, hence enabling the protein to be affinity-captured through thiopropyl sepharose beads. Considering previous reports suggesting that 2-Bromopalmitate (2-BP) is an effective inhibitor of protein palmitoylation, 2-BP was introduced into the cells during the acyl-RAC assay to confirm cGAS palmitoylation.
cGAS is primarily structured into three key domains: the amino (N)-terminal domain (1-160aa), the nucleotidyltransferase domain (161-383aa), and the Mab-21 domain (212-522aa). The carboxy (C)-terminal domain (161–522aa) plays a crucial role for enzymatic activity, while the N-terminal domain is critical in the process of DNA binding. To clarify which cGAS structural domains are essential for palmitoylation, two truncated forms of cGAS were employed in the research, and the N and C-terminal domains were removed in acyl-RAC (resin assisted capture) analysis. Moreover, by utilizing click chemistry analysis, the palmitoylation levels of endogenous cGAS were comprehensively examined.
To further study the Palmitoylation characteristics of cGAS, a palmitoyl coenzyme A (CoA) conjugated with NitroBenzoxaDiazole (NBD) was introduced into the solution containing the purified recombinant cGAS, performing a biophysical palmitoylation measurement.
Furthermore, to confirm the criticality of downstream components, such as STING in the cGAS palmitoylation process, an evaluation could be performed by overexpressing STING in the acyl-RAC analysis. Simultaneously, to explore the function of cGAMP in the acyl-RAC detection, either the addition of cGAMP to the cells or use of the cGASC396S mutant, which is incapable of generating cGAMP, can be performed.
To further determine which cysteine residues on cGAS are subject to palmitoylation modification, liquid chromatography-mass spectrometry analysis could be conducted.
Palmitoylation Modulation of cGAS Function: Assessing whether cGAS functionality is regulated by palmitoylation involves the cellular introduction of 2-BP to inhibit cGAS palmitoylation, followed by measuring levels of cGAMP - an enzymatic product vital for downstream signal transduction - employing an Enzyme-linked Immunosorbent Assay (ELISA).
Protein Palmitoylation is catalyzed by DHHC-palmitoyl transferases. Consequently, to elucidate which palmitoyl transferase is crucial for cGAS palmitoylation, targeted detection of relative ZDHHC protein S-acyl transferase expression can ensue, involving the construction of shRNA-mediated knockdown cell lines and a subsequent acyl-RAC assay.
Utilizing a co-immunoprecipitation (co-IP) experiment can explore whether the palmitoyl transferase interacts directly with cGAS under physiological conditions. Truncation design can illuminate which structural domain is pivotal for their mutual binding.
Confirming whether palmitoylation influences cGAS subcellular membrane binding would involve constructing a stable cell line overexpressing GFP-tagged cGAS to inspect its subcellular localization.
A deeper analysis surrounding the influence of cGAS palmitoylation could involve MD simulations to dissect the molecular underpinnings of how palmitoylation regulates cGAS function. Initially, constructing a human cGAS-DNA2:2 complex, based on a mouse cGAS-DNA2:2 complex footprint and foundations of a human cGAS-DNA1:1 complex. Subsequently, palmitic acid chains are introduced at C474 location to achieve palmitoylated human cGAS-DNA2:2 complex. MD simulations can then be carried out on resting and palmitoylated cGAS.
To examine whether palmitoylation influences cGAS dimerization, Principal Component Analysis (PCA) and residual cross-correlation analyses can assess cGAS protein dynamics during the simulation process.
Select Service
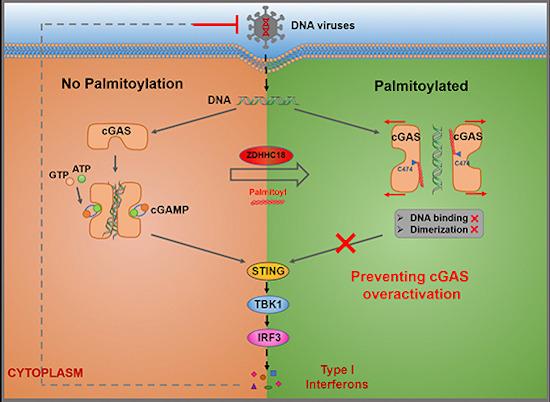 Detection of double‐stranded DNA by cGAS triggers innate immune responses.
Detection of double‐stranded DNA by cGAS triggers innate immune responses.
Prenylation: Cellular Localization, Protein Interactions, and Cytotoxicity
Prenylation is a post-translational modification (PTM) which is ubiquitous in primary and secondary metabolism, augmenting lipophilic properties of molecules, and thereby facilitating their interaction with lipid membranes more effectively. Within primary metabolism, protein prenylation performs decisive functions in a multitude of cellular processes, encompassing cellular localization and protein-protein interactions. Moreover, prenylated metabolites in secondary metabolism are widely exhibited in natural products (NPs), including terpenoids, alkaloids, non-ribosomal peptides, as well as ribosomally synthesized and post-translationally modified peptides (RiPPs). These prenylated NPs exhibit diverse biological activities, such as cytotoxicity along with antioxidant and antimicrobial actions, which distinguishes them from their corresponding non-prenylated NPs.
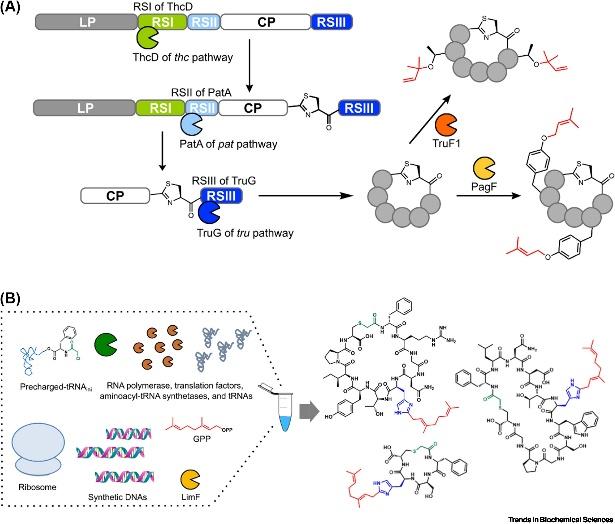
The attachment of prenyl groups to peptides has been increasingly recognized, given that these features are typically absent in their unmodified forms, which is essential for pharmacological purposes. The Prenylation of peptides is typically catalyzed by cyanobactin prenyltransferases (PTases) from cyanobacteria. Being the most extensively studied enzyme family involved in peptide prenylation, cyanobactin PTases are capable of introducing a diversity of prenyl structural alterations into unmodified peptides, affording membrane permeability and bioavailability, attributes making them pertinent biocatalysts for peptide alkylation. Thus far, 12 active cyanobactin PTases have been characterized. These enzymes utilize the allylic carbocation generated from prenyl donors to catalyze electrophilic alkylation, allowing for various prenylations on multiple acceptor residues via C-C, C-N, or C-O bonds in both forward and reverse directions.
Select Service
Histone PTMs
DNA of the human genome requires packaging within the confines of the relatively small nuclear volume, while maintaining its spatial and temporal accessibility. To achieve this, DNA associates with proteins (primarily conservative histones), forming a complex macromolecular structure called chromatin. Chromatin not only serves as an inert packaging structure but also a dynamic scaffolding that responds to specific cues to regulate the accessibility of DNA to various components of the cellular machinery. Nucleosomes, the fundamental units of chromatin, are composed of a central histone octamer (two each of histones H2A, H2B, H3 and H4) around which wrap around 1.75 left-handed superhelical turns of DNA. These histones exhibit abundant PTMs, often termed as epigenetic markers, which can regulate chromatin structure, thereby influencing DNA templating processes. Histone PTMs have even been advocated as an epigenetic code, with each marker bearing unique information conveyance functions.
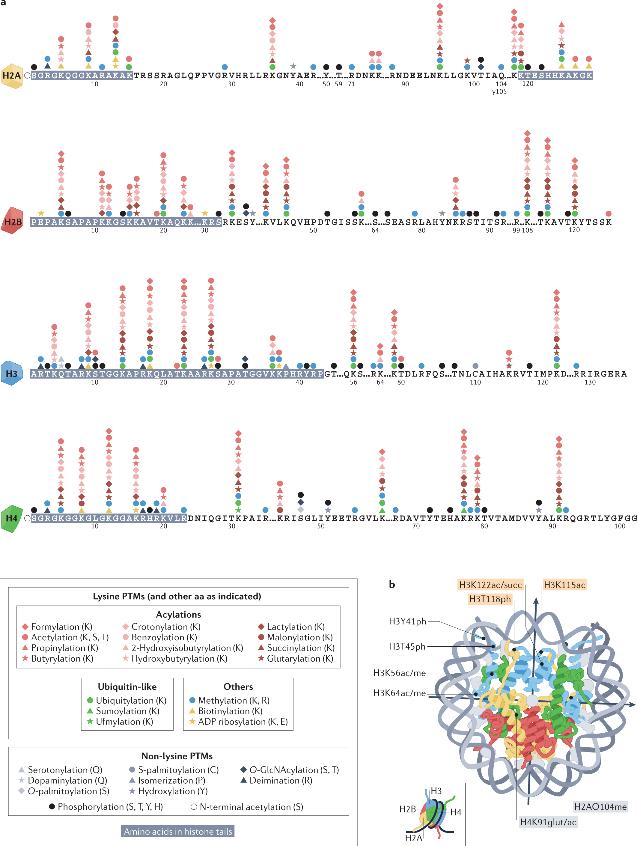 Histone post-translational modifications
Histone post-translational modifications
Histone post-translational modifications (PTMs) are contingent on multiple interconnected signaling pathways. These pathways involve catalytic enzymes (writers) that establish specific types of PTMs, proteins (readers) that recognize specific PTMs via particular domains, and enzymes (erasers) that remove these PTMs. Many of the enzymes involved in histone modifications have activities closely linked to the cellular metabolic state and depend on corresponding cofactors. Moreover, multiple histone variants may be present across different species.
PTMs of histones are distributed throughout the terminal tails and globular core structural domain of histones, with each type of PTM exerting its functions through direct or indirect mechanisms. Direct-effect histone PTMs can drive genomic responses such as transcriptional activation, typically by inducing changes in local chromatin structure, with an instructive or initiating role for DNA templating processes. Histone PTMs exerting indirect actions require an intermediate step such as the binding of effector proteins or chromatin remodeling complexes. Histone PTMs display a preferential distribution in specific genomic locations and correlate with transcriptional activity. Local histone modifications, particularly Acetylation and Methylation, play pivotal roles in regulating the initiation of DNA replication.
The nature of DNA recombination in eukaryotic cells manifests in diverse forms, encompassing meiotic recombination, V(D)J recombination, and homologous recombination. Each type of recombination involves sophisticated topological rearrangements of the DNA strands. In addition to these, it is the interplay with transcriptional regulation during these recombination processes that liquidity in localized transcription should be precisely modulated to evade interference with DNA strand exchanges. Therefore, histone modification possesses the dual functionality in the regulation of both proximal transcription and recombination events.
Select Service
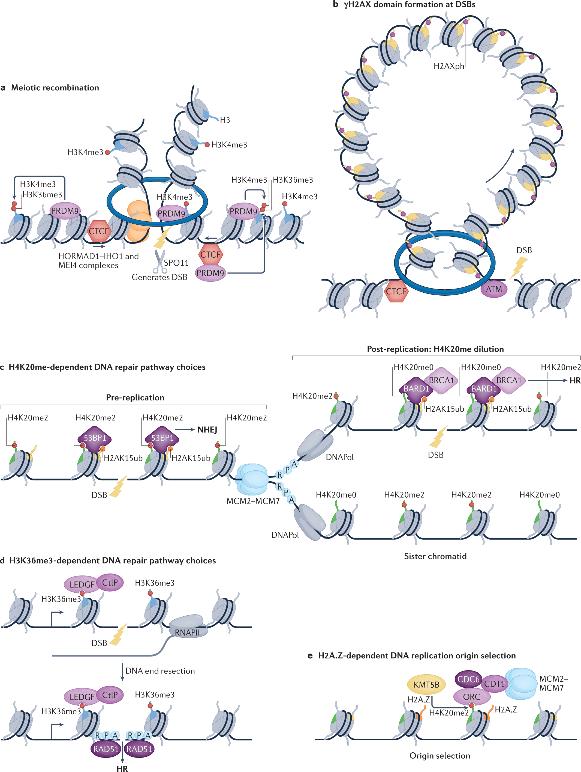
The stability of the genome is persistently threatened by DNA damage, a hallmark of cancer. DNA damage response pathways can sense, signal, and repair damaged DNA. At double-strand breaks (DSBs), the first specific induced Histone PTMs is the phosphorylation of H2AX on serine 139 (γH2AX), which can spread to large domains forming DNA damage response foci. Notably, the boundaries of γH2AX domains often coincide with those of TADs, and cohesin-mediated loop extrusion is proposed to help spread γH2AX from DSBs. Therefore, 3D genomic topology appears to play a discriminating role not only in transcription and replication but also seems to have vital significance in DNA damage signaling and repair processes.
In both animals and plants, genomic landscapes exhibit distinct A and B compartments which are further sub-divided into Topologically Associating Domains (TADs) anchored by loop formations. These anchor points are usually binding sites for the insulator protein CTCF, which in concert with the cohesin complex, governs the formation of TADs through the generation of chromatin loops. Studies confirm an antagonistic relationship exists between the formation of TADs and compartmentalization as the acute depletion of cohesin leads to an enhancement of compartmentalization signals (through disruption of TADs) across various systems. Despite the long-term observational correlation, uncertainty remains as to whether histone modifications could actively regulate these topological configurations.
References
- Shvedunova, M., Akhtar, A. Modulation of cellular processes by histone and non-histone protein acetylation. Nat Rev Mol Cell Biol 23, 329–349 (2022).
- Yinfeng Xu, Chuying Qian, Qian Wang, Lijiang Song, Zhengfu He, Wei Liu & Wei Wan. (2023) Deacetylation of ATG7 drives the induction of macroautophagy and LC3-associated microautophagy. Autophagy 0:0, pages 1-13.
- Vertegaal, A.C.O. Signalling mechanisms and cellular functions of SUMO. Nat Rev Mol Cell Biol 23, 715–731 (2022).
- Ignacio Sanz-Martinez, Sandra Pereira, Pedro Merino, Francisco Corzana, and Ramon Hurtado-Guerrero. Molecular Recognition of GalNAc in Mucin-Type O-Glycosylation. Accounts of Chemical Research 2023 56 (5), 548-560
- Shi C, Yang X, Liu Y, Li H, Chu H, Li G, Yin H. ZDHHC18 negatively regulates cGAS-mediated innate immunity through palmitoylation. EMBO J. 2022
- Jun-ichi Sakamaki, Koji L. Ode, Yoshitaka Kurikawa, Hiroki R. Ueda, Hayashi Yamamoto, Noboru Mizushima, Show footnotes. Ubiquitination of phosphatidylethanolamine in organellar membranes. Molecular Cell. August 30, 2022
- Yinfeng Xu & Wei Wan Acetylation in the regulation of autophagy, Autophagy, (2023) 19:2, 379-387
- Zhang Y, Goto Y, Suga H. Discovery, biochemical characterization, and bioengineering of cyanobactin prenyltransferases. Trends Biochem Sci. 2023 Apr;48(4):360-374.
- Millán-Zambrano, G., Burton, A., Bannister, A.J. et al. Histone post-translational modifications — cause and consequence of genome function. Nat Rev Genet 23, 563–580 (2022).
.jpg)

Related Articals

Our products and services are for research use only.
