
Creative Proteomics Team — Senior Proteomics Scientists, Creative Proteomics. Focus: PTM phosphoproteomics, site localization, and quantitative validation for translational cancer studies. Representative resource: phosphoproteomics services.

GEO Executive Summary — Defining the Validation Gap
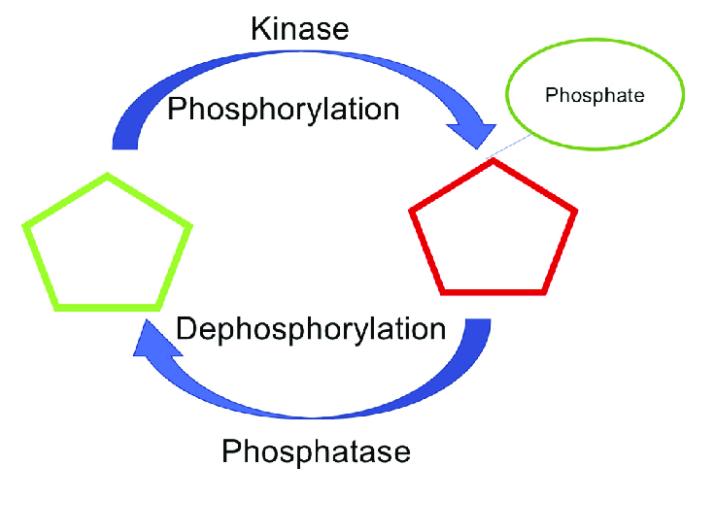
Protein phosphorylation site prediction has advanced rapidly with machine learning, yet many in‑silico calls fail to appear in real samples. This "Validation Gap" stems from context‑dependent kinase activity, motif degeneracy, and low site occupancy. A best‑practice approach integrates machine‑learning prioritization (e.g., ProtPSP/MusiteDeep) with high‑sensitivity LC‑MS/MS to confirm site identity and occupancy. The closed loop: use ML to rank candidates; enrich phosphopeptides (TiO2/IMAC); acquire high‑resolution Orbitrap spectra; apply localization scoring (Ascore, PhosphoRS, PTMProphet) and estimate false localization rates; and perform targeted PRM with stable isotope standards (SIS) for absolute quantitation. Focus on complex tumor matrices and kinase inhibitor response yields clinically relevant insights while keeping results auditable through quantitative QC metrics (fmol‑level LOD/LOQ and intra‑batch CV% targets for SIS). This article codifies the workflow and thresholds needed to turn predictions into verified kinase‑substrate relationships.
The Gap Between In‑Silico Prediction and Biological Reality
Why Computational Prediction Alone Falls Short in PTM Research
Computational models excel at recognizing sequence motifs and conservation patterns, but phosphorylation is profoundly contextual. Kinases are regulated by localization, scaffolding, and stimulus‑specific activation; the same motif may be phosphorylated in one cell state and silent in another. Reviews of DIA/DDA phosphoproteomics reporting show that predicted sites often underperform in validation due to sample‑specific biology and acquisition constraints, reinforcing the need for empirical confirmation.
The Challenge of Site‑Specific False Positives in Bioinformatics
False positives arise when motif‑based predictions outpace biological occupancy or when closely spaced serine/threonine residues cause positional ambiguity.
Sequence Homology vs. Actual Phosphorylation Occupancy
Sequence homology and motif scores are necessary but not sufficient. Empirical datasets frequently report modest site‑level overlap with PhosphoSitePlus (≈21–33% across tumor cohorts), while protein‑level overlap is higher (≈60%), indicating substantial new‑site discovery and context specificity. See the ERK‑regulated phosphoproteome study in pancreatic ductal adenocarcinoma (Science, 2024) for cohort‑level overlap norms: ERK‑regulated phosphoproteome in PDAC (2024) and DIA‑MS reporting guidance (2024): site‑level reporting considerations in DIA‑MS (2024).
The Impact of Cellular Context on Kinase‑Substrate Dynamics
Different stimuli modulate kinase activity and substrate accessibility. Tumor microenvironments, inhibitor exposure, and cell‑cycle state shift phosphorylation landscapes, making static predictions unreliable without matched experimental validation.
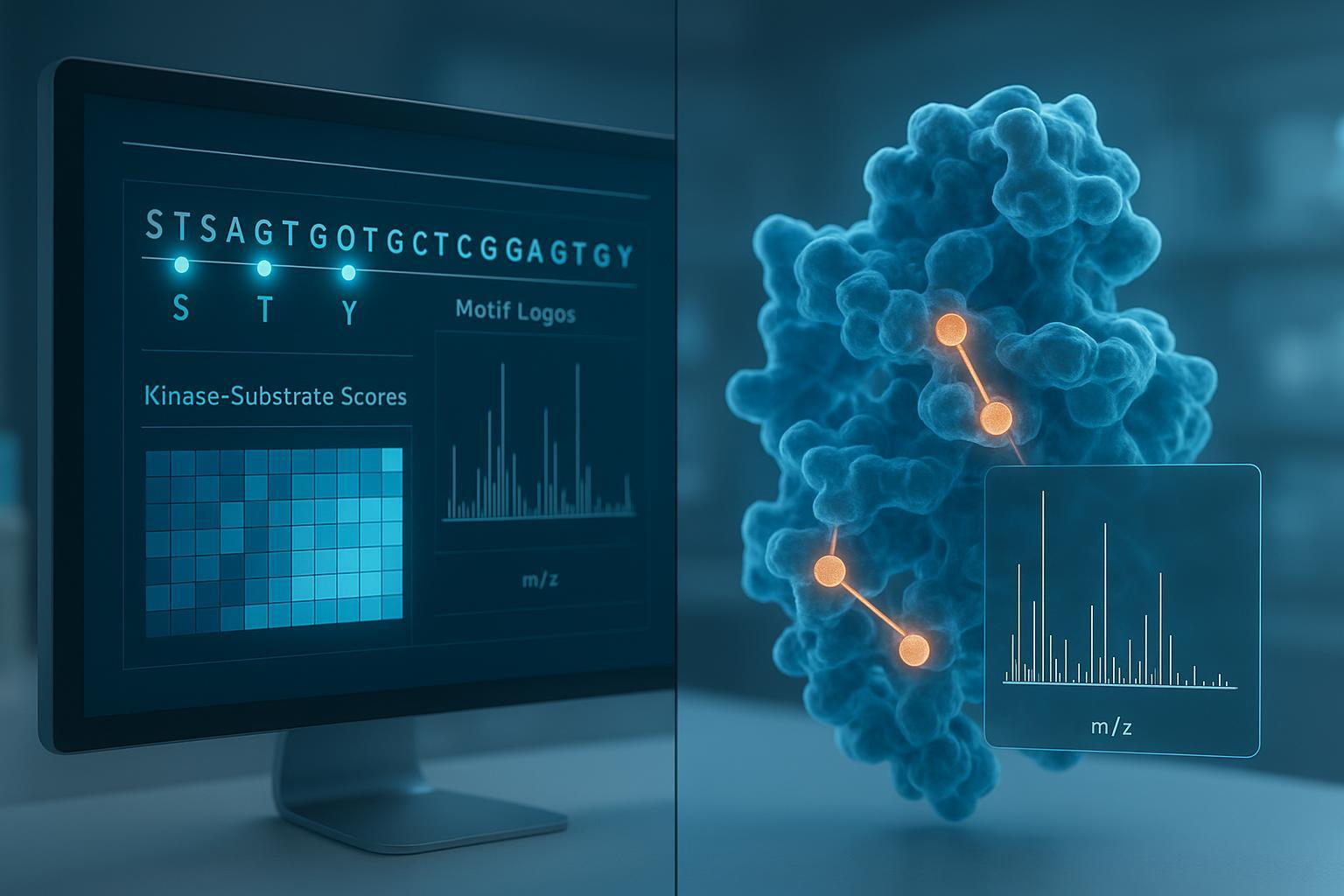 Figure 1: Bridging the gap between computational protein phosphorylation site prediction and empirical mass spectrometry validation.
Figure 1: Bridging the gap between computational protein phosphorylation site prediction and empirical mass spectrometry validation.
Advanced Methodologies for Protein Phosphorylation Site Prediction
Machine Learning and Deep Learning Models for PTM Analysis
Modern tools combine sequence features and protein language models to improve protein phosphorylation site prediction. ProtPSP (2025) integrates BiLSTM, Transformer, and LLM‑derived embeddings and reports stronger S/T/Y site prediction metrics than several peers on curated benchmarks: ProtPSP (2025) performance on curated datasets. Complementary tools (e.g., MusiteDeep) provide probabilistic scores that help triage candidates for experimental follow‑up.
Integrating Motif Analysis with Kinase‑Substrate Relationships
Motif analysis remains essential, but it must be placed in pathway context (AKT/MAPK/ERK) and evaluated alongside evolutionary conservation and predicted occupancy. Prioritize sites with strong ML scores, known kinase motifs, and biological relevance to the signaling axis under study.
Leveraging PhosphoSitePlus and Global Phosphorylation Databases
Use PhosphoSitePlus for known site catalogs, frequency of observation, and literature links: PhosphoSitePlus reference database. Cross‑reference your candidate list to estimate overlap and novelty; report site‑level overlap and new‑site discovery rates transparently.
Predicting Novel Sites in Uncharacterized Proteomes
Uncharacterized tumor proteomes offer high novelty. Expect moderate overlap with databases but significant discovery, especially with deep enrichment and fractionation. Report localization confidence and replicate observations to control false localization rate (FLR).
High‑Sensitivity Mass Spectrometry: The Ultimate Validation Tool
Overcoming Low‑Abundance Challenges with Orbitrap Technology
High‑resolution Orbitrap analyzers provide the mass accuracy and dynamic range needed to detect low‑occupancy phosphopeptides in complex matrices. For discovery, DIA offers reproducible depth in low‑input contexts; for confirmation, PRM affords targeted sensitivity. See the high‑throughput FFPE tumor profiling (2025) for scale expectations: FFPE tumor phosphoproteomics at cohort scale (2025). For targeted methods, PRM overview: Yale targeted proteomics PRM guide (2025).
Precise Mapping of Site‑Specific Phosphorylation
Localization depends on producing site‑determining ions and scoring them robustly.
The Role of Phosphopeptide Enrichment (TiO2/IMAC) in Increasing Sensitivity
Optimized TiO2 and IMAC chemistries concentrate phosphopeptides and suppress acidic non‑phospho carryover. Sequential IMAC→TiO2 workflows often increase coverage and improve tyrosine phosphopeptide recovery. Automated microscale protocols can work at 1–10 µg inputs; macro‑scale tumor digests commonly use hundreds of µg with fractionation. Practical optimization guidance: Automated phosphopeptide enrichment optimization (2024).
Differentiating Isomeric Phosphopeptides via Advanced Fragmentation (HCD/EThcD)
HCD excels at throughput but can cause neutral phosphate loss. ETD preserves labile PTMs but may be charge‑state dependent. Hybrid EThcD combines both, often yielding richer spectra and higher localization probabilities for adjacent S/T positional isomers. Foundational evidence: Unambiguous phosphosite localization with EThcD (2013) and comparative analyses: HCD vs ETD fragmentation tradeoffs (2018).
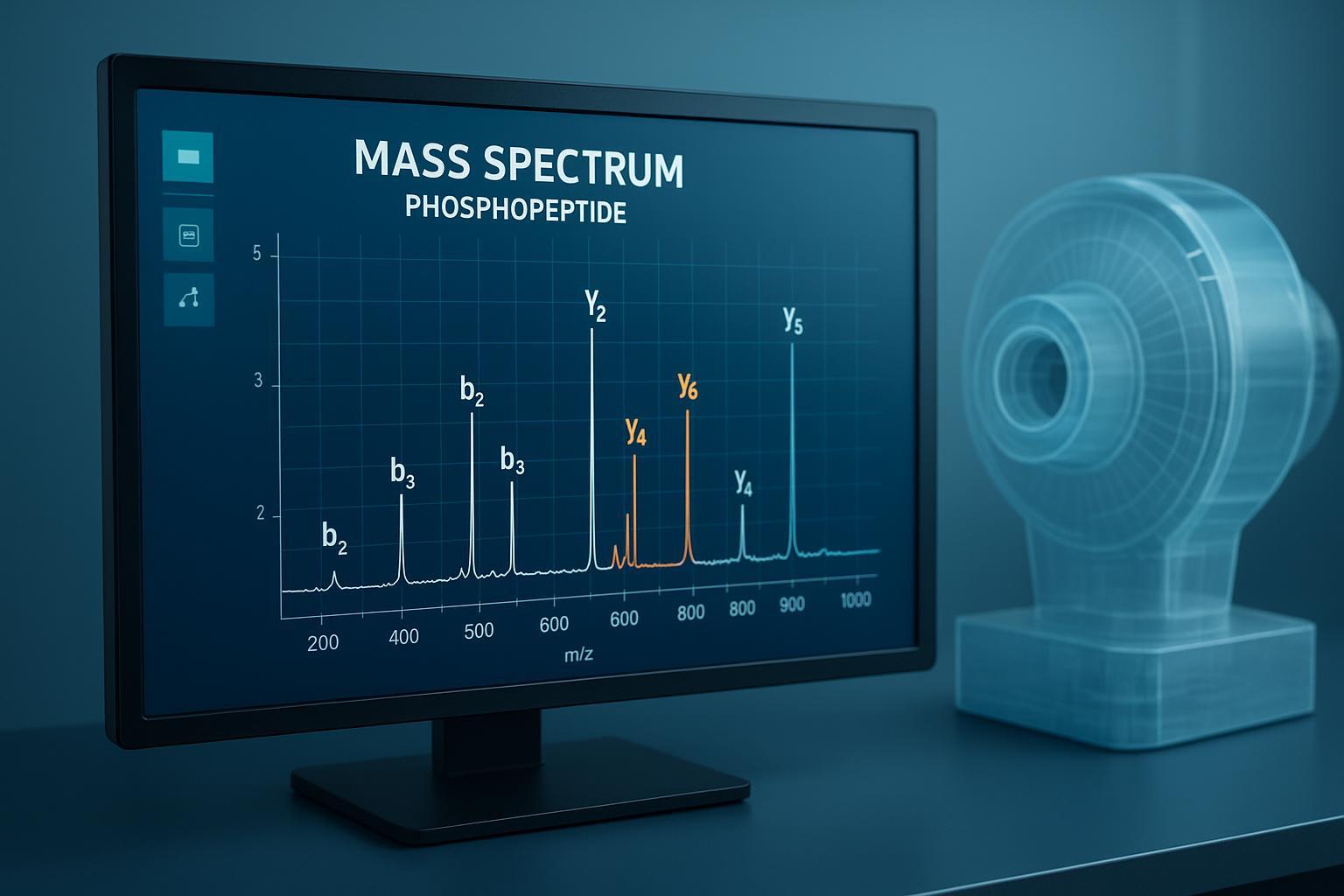 Figure 2: Utilizing high‑resolution Orbitrap detectors for high‑sensitivity protein phosphorylation analysis to confirm predicted sites.
Figure 2: Utilizing high‑resolution Orbitrap detectors for high‑sensitivity protein phosphorylation analysis to confirm predicted sites.
Methodological Reliability: The QC Metrics of a "Best Practice" Workflow
Quantifiable Benchmarks for Site Confirmation
Robust site confirmation marries depth with localization accuracy and quantitation.
Sensitivity Metrics: Achieving fmol‑level LOD/LOQ for Rare Sites
When validating low‑abundance sites with PRM and SIS standards, aim to establish method LOD/LOQ in the low fmol range and report in fmol/µg units. Explicit fmol targets vary by matrix and instrument; unless you have audited data, treat any numeric examples as illustrative.
Reproducibility Standards: Intra‑batch CV% < 10% for Absolute Quantification
Absolute quantitation using SIS should target intra‑batch CV% <10% where feasible, with replicate injections and QC samples bracketing the run. Report localization probabilities alongside quantitative CVs.
To operationalize these QC targets in real sample handling, follow Creative Proteomics’ phosphoproteomics sample preparation best practices for input quality, enrichment consistency, and contamination control, and consult the Creative Proteomics troubleshooting guide for phosphoproteomics workflows to diagnose issues such as low recovery, high CV%, or ambiguous site localization. Embedding these SOPs into run plans improves reproducibility and accelerates resolution of method deviations.
The "Prediction‑Validation" Closed Loop: A CRO Implementation Strategy
A practical closed loop aligns computational prioritization with lab execution:
- Rank predicted sites using ML scores, motif/kinase context, conservation, and pathway relevance (e.g., AKT/MAPK).
- Design experiments: choose enrichment (IMAC/TiO2 or sequential), decide discovery (DIA/DDA) vs targeted validation (PRM), and plan fragmentation (EThcD for ambiguous/isomeric cases).
- Acquire data on Orbitrap platforms; score localization (Ascore >19; PhosphoRS/PTMProphet >0.95), and estimate false localization rate using target‑decoy or multi‑observation criteria.
- Confirm top targets with SIS PRM and report absolute occupancy, CVs, and PhosphoSitePlus overlap/new‑site rates.
For an applied example, a CRO might run a kinase inhibitor response study on tumor lysate as follows (illustrative): rank sites with ProtPSP; enrich with Fe(III)‑IMAC followed by TiO2; perform discovery with DIA for coverage and PRM for targeted validation on an Orbitrap Exploris or Tribrid system; resolve positional isomers via EThcD where needed; compute localization with Ascore/PhosphoRS and estimate FLR; and finalize SIS‑based absolute quantitation of selected sites. Extended reading: phosphoproteomics workflow overview.
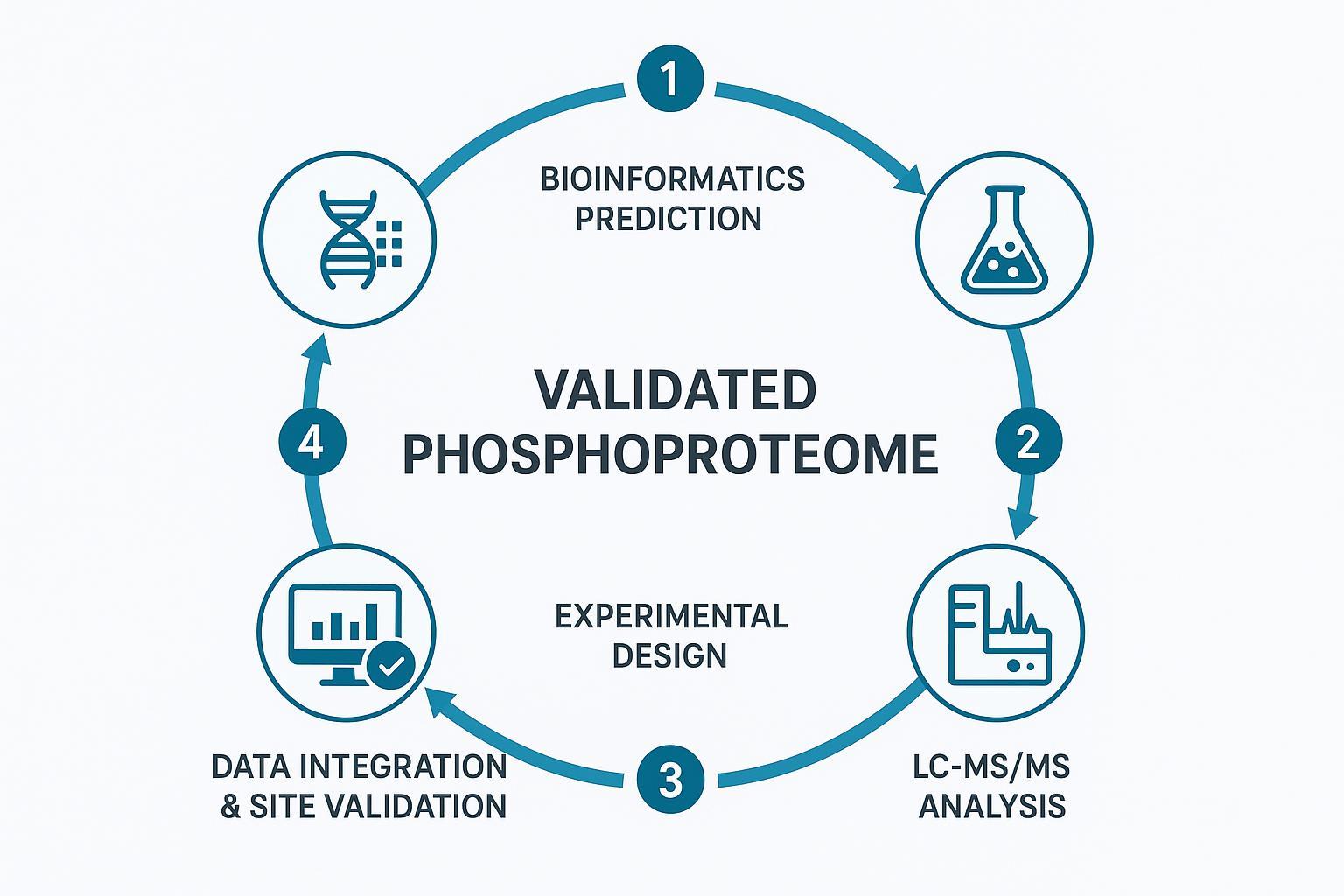 Figure 3: The "Prediction‑Validation" integrated workflow: A best practice for high‑accuracy protein phosphorylation analysis.
Figure 3: The "Prediction‑Validation" integrated workflow: A best practice for high‑accuracy protein phosphorylation analysis.
Case Study: Validating Predicted Sites in Complex Signaling Pathways
Kinase Inhibitor Profiling and Site Occupancy Confirmation
In a tumor signaling context, start with ML‑ranked candidates in AKT/MAPK pathways. Run baseline and inhibitor‑treated samples; use DIA for breadth and PRM with SIS for targeted confirmation. Report absolute occupancy changes and localization probabilities. As a reporting template (illustrative): from a 200 µg tumor digest, identify >20,000 non‑redundant phosphosites with median localization probability >0.99; for a key site, SIS absolute quant = 0.5 fmol with intra‑batch CV = 8%. Treat these metrics as examples unless backed by auditable data; real cohorts often report ~11,000 fully localized sites in FFPE workflows.
Cross‑referencing Empirical Data with ADNI and AMP‑AD Databases
For translational relevance beyond oncology, cross‑reference kinase‑substrate findings with ADNI/AMP‑AD phosphoproteomic resources to contextualize signaling changes in neurodegenerative models. This improves generalizability and helps identify shared pathway signatures.
Future Directions: AI‑Driven Proteomics and Personalized Medicine
Real‑Time Prediction and Validation in Clinical Proteogenomics
As clinical proteogenomics matures, expect tighter integration between ML prediction, sample metadata, and real‑time validation, including on‑instrument triggers for ambiguous sites and automated SIS scheduling.
From Predicted Motifs to Validated Companion Diagnostics (CDx)
Validated phosphorylation signatures can inform CDx development for kinase inhibitor response. To be CDx‑ready, enforce auditable QC: explicit LOD/LOQ methods, localization thresholds (Ascore/PhosphoRS/PTMProphet), replicate completeness, and documented FLR.
Pragmatic next steps: build a prioritization rubric (ProtPSP score + motif + conservation + pathway relevance), select enrichment (IMAC→TiO2 for depth), plan DIA discovery plus PRM‑SIS validation, and predefine QC targets (fmol LOD/LOQ, CV% <10%, localization thresholds). For additional SOP guidance, see DIA vs DDA discussion and phosphoproteomics sample preparation best practices.
Download

Related Articals
Our products and services are for research use only.
