
Phosphorylation–ubiquitination crosstalk sits at the control panel of protein fate. Kinases can install phospho-degrons that recruit E3 ligases, triggering site-specific ubiquitination, signal termination, and often degradation by the proteasome. Conversely, lysine competition by acetylation or SUMOylation can delay or divert ubiquitination, shaping proteostasis and signaling duration. For translational projects, the implication is practical: if you want to measure how a pathway turns off, or why a substrate's half-life collapses under a kinase inhibitor, you need a unified, quantitative view of both phosphorylation and ubiquitination from the same biological system—ideally with site-level confidence, robust quantitation, and auditable QC.
This ultimate guide teaches an end-to-end methodology for co-analyzing these modifications with an emphasis on phospho-degrons. You'll find sequential enrichment options, DIA/DDA acquisition choices, multiplexed quant strategies, occupancy and stoichiometry calculations, and verification via PRM and kinetics—plus the QC targets that keep reviewers and collaborators confident.
Key takeaways
- Phosphorylation–ubiquitination crosstalk often hinges on phospho-degrons that recruit E3 ligases to mark substrates for turnover.
- Combine phospho enrichment with diGly ubiquitin-remnant enrichment and pair with DIA for cohort-level reproducibility.
- Use clear QC benchmarks: ≤1% peptide FDR, defined site-localization probability, and intra-batch median CV near or below 15% when workflows are well controlled.
Beyond single modifications – why phosphorylation–ubiquitination crosstalk matters
Phosphorylation can encode timing and direction. When specific serines or threonines are phosphorylated in degron motifs, WD40-domain E3 receptors bind with high affinity, catalyzing ubiquitination that commits a substrate to degradation. Canonical examples include β-catenin and IκB via SCF β-TrCP, and cyclin E and c-Myc via FBXW7.
Interplay between phosphorylation and ubiquitination
At the motif level, β-TrCP recognizes a diphosphorylated DSGxx(X)pS motif; structural work on β-catenin phosphopeptides showed how phosphorylated serines coordinate binding and enable ubiquitination, as detailed in the 2004 structural analysis by Watanabe and colleagues in PNAS. Earlier biochemical evidence demonstrated that phosphorylation enables β-TrCP binding to the DSG motif, as reported by Lassot and colleagues in 2001 in Molecular and Cellular Biology. For FBXW7, dually phosphorylated CPDs enable high-affinity binding; recent work revealed two diphosphorylated degrons in Myc that together control stability, refining earlier cyclin E paradigms, summarized in 2022 by Welcker and colleagues in Science Advances.
Signaling vs degradation – how PTMs dictate protein fate
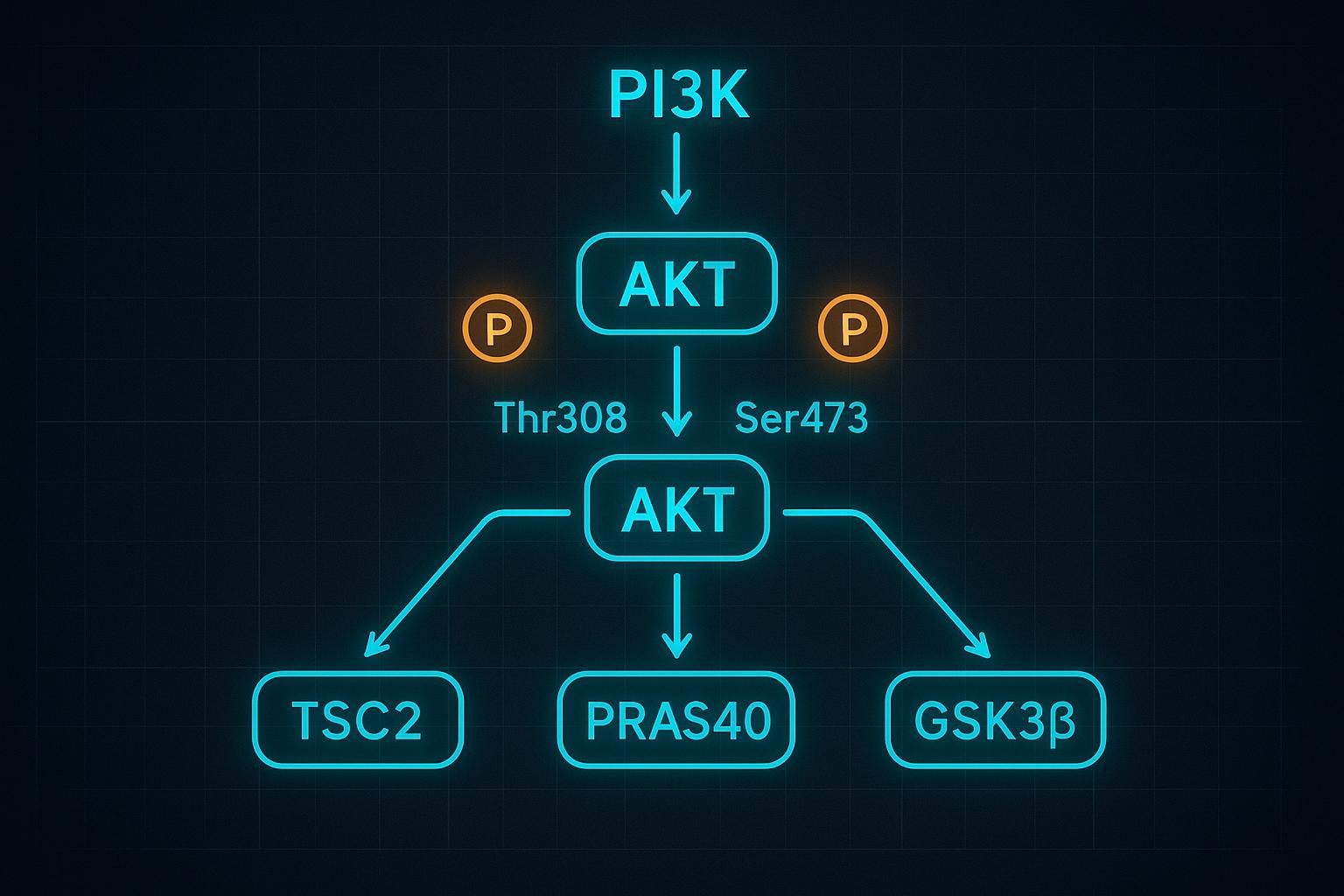
Whether phosphorylation signals activity, recruits readers, or flags a substrate for destruction depends on context. Glycogen synthase kinase 3 beta often completes phospho-degrons; AKT inhibits GSK3β through pS9, indirectly stabilizing many FBXW7 and β-TrCP substrates. Meanwhile, the balance at lysines matters: acetylation and SUMOylation can occupy the same residues or adjacent motifs, altering ubiquitin chain initiation or extension and shifting a protein's half-life.
Phosphorylation-dependent degron motifs
SCF β-TrCP phospho-degrons often follow DpSGxx(X)pS variants, while FBXW7 binds disordered, diphosphorylated CPDs typically primed by CDKs and extended by GSK3β. Consensus and substrate inventories have expanded across oncogenic and signaling pathways.
Competition at lysine residues – acetylation, ubiquitination, sumoylation
Acetylation can block ubiquitin conjugation at target lysines, delaying degradation; SUMOylation may either compete directly or create SUMO-dependent ubiquitination signals via SUMO-targeted ubiquitin ligases. Interpreting crosstalk therefore requires measuring multiple PTMs in parallel or in matched aliquots.
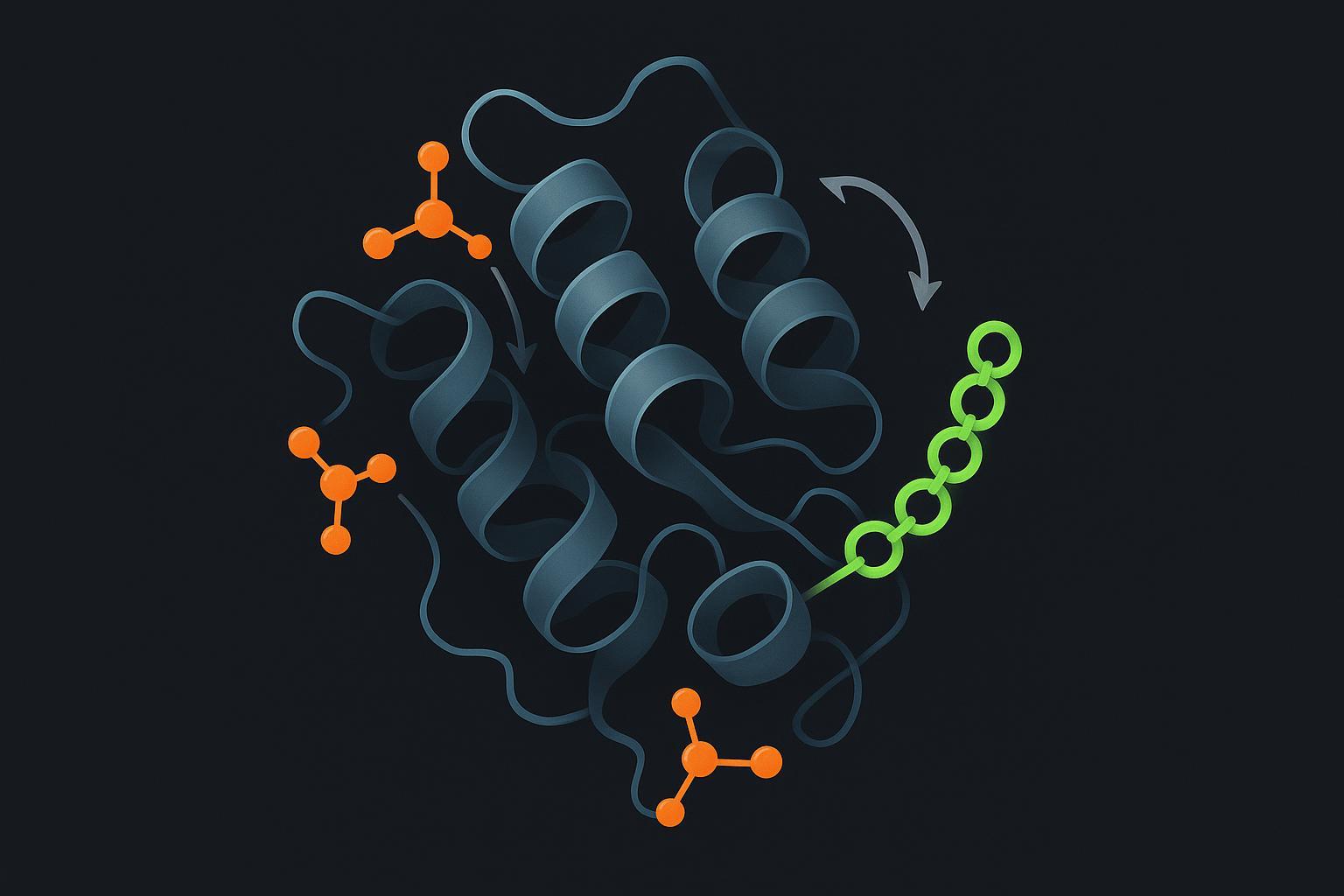
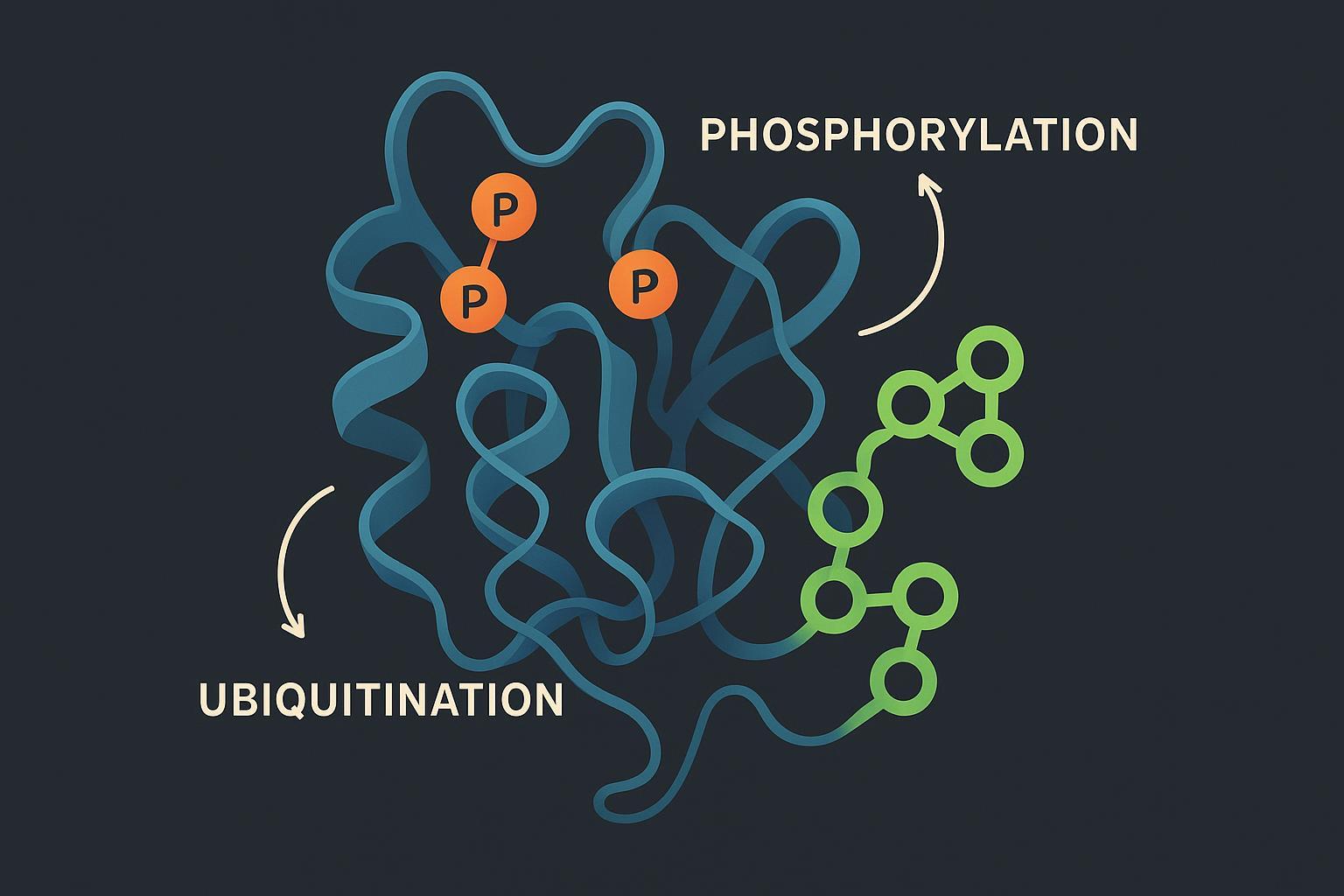 Figure 1: Molecular crosstalk within the phosphorylation–ubiquitination axis regulating protein stability.
Figure 1: Molecular crosstalk within the phosphorylation–ubiquitination axis regulating protein stability.
Methodology – integrated co-analysis of dual PTMs
The practical goal is quantitative, site-resolved measurement of phospho and ubiquitin signals with auditable QC. In most studies, split matched aliquots after digestion: one for phospho enrichment and one for diGly enrichment. True same-aliquot sequential approaches exist but may trade sensitivity.
Sequential enrichment strategies – SIMAC and SMOAC
SIMAC or SMOAC extends phosphoproteome depth by combining IMAC and TiO2 sequentially, capturing complementary mono- and multi-phosphorylated peptides. Comparative and protocol resources show complementary capture between Fe-IMAC and TiO2 and improved coverage via sequential strategies, for example Yue and colleagues' 2015 comparison in Proteomics and the Rutgers SMOAC protocol.
Independent benchmarks from peer-reviewed groups favor SMOAC (or SMOAC-like metal-oxide sequencing) plus diGly workflows for dual-PTM projects. Large protocol/benchmark studies (Broad Institute Nat. Protoc., 2020) and multi-center proteome work show diGly yields the highest K-ε-GG specificity while SMOAC gives superior phospho depth and orthogonality; hybrids or SMOAC flow‑through → diGly raise total co-identified sites versus SIMAC alone (Nat. Protoc. 2020; Mol. Cell Proteomics 2023).
Ubiquitin-remnant diGly enrichment and caveats
Trypsin digestion of ubiquitinated lysines leaves a K-ε-GG remnant. Anti–K-ε-GG antibodies capture these peptides with high specificity; magnetic bead formats improve handling at lower inputs. Caveats include cross-reactivity with NEDD8 and ISG15 and yield losses without proper antibody crosslinking and buffer exchange. Vendor and peer-reviewed descriptions outline performance and specificity, including the CST PTMScan K-ε-GG kit overview and cohort-scale outcomes.
Optimizing TMT multiplexing for phospho and diGly quantitation
Use SPS-MS3 or TurboTMT on Orbitrap Tribrids to mitigate ratio compression in complex PTM samples. Reviews and benchmarks detail labeling efficiency targets and MS3 benefits; for example, Weiner and colleagues' 2022 analysis in Molecular & Cellular Proteomics.
Sensitivity metrics – bridging the stoichiometry gap
Phosphorylation can be high-occupancy on some sites, while site-specific ubiquitination is often sparse. Prefer DIA for cohort-scale studies to reduce missingness and improve sensitivity to low-abundance diGly peptides. A benchmark in ubiquitinome profiling is Steger and colleagues' 2021 time-resolved work in Nature Communications. Use heavy standards: Ub-AQUA peptides for absolute quantitation of linkages and select sites, and Ub-PSAQ heavy proteins pre-lysis to correct recovery, originally described by Kaiser and colleagues in 2011 in Nature Methods.
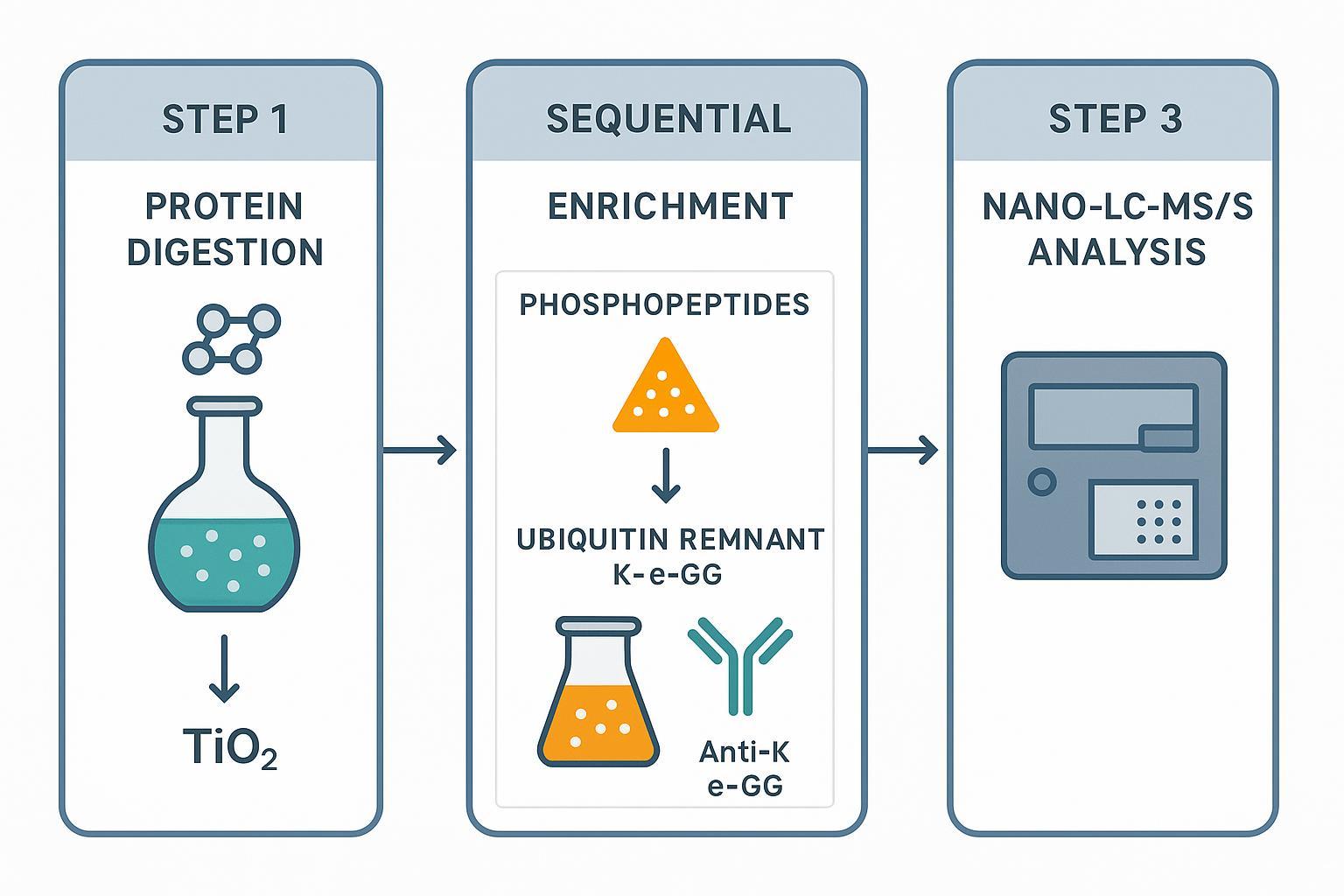 Figure 2: Integrated workflow for the simultaneous quantitative analysis of phosphorylation and ubiquitination.
Figure 2: Integrated workflow for the simultaneous quantitative analysis of phosphorylation and ubiquitination.
Acquisition and quantitation – Orbitrap DDA, DIA, and PRM
Choosing DDA vs DIA for discovery and cohorts
DDA excels in discovery with deep fractionation, but missing values can challenge cohort comparisons. DIA improves data completeness and reproducibility for both phospho and ubiquitinomics, enabling consistent quantitation of thousands of sites across large cohorts; library-free DIA is viable on modern platforms. For a benchmark in ubiquitinome profiling, see the large-scale, time-resolved DIA study noted above.
For an acquisition strategy comparison in phosphoproteomics, a neutral resource outlines DIA vs DDA trade-offs and cohort reproducibility: phosphoproteomics DIA vs DDA.
PRM and absolute quantitation with AQUA and PSAQ
PRM on Orbitrap instruments offers sensitive, selective quantitation of targeted phospho and diGly peptides. Schedule retention windows and use 30k–60k MS2 resolution with 0.7–1.2 m/z isolation. For phosphorylation occupancy, synthesize heavy standards for both modified and unmodified peptides and compute occupancy as P/(P+U). A tutorial-style application is described by Stein and colleagues in 2021 in Cell Reports Methods. For absolute ubiquitination, spike Ub-AQUA heavy peptides post-digestion to quantify site or chain types; for absolute pools and recovery correction, spike Ub-PSAQ heavy ubiquitin proteins pre-lysis, then deconjugate with DUBs as outlined in Nature Methods.
Reliability and QC – quantifying the interaction network
Site-specific occupancy and stoichiometry calculations
Two practical calculations:
Phosphorylation occupancy: quantify the phosphorylated peptide (P) and its unmodified counterpart (U) by PRM with heavy standards; compute occupancy = P/(P+U). Ensure co-elution and linear response within calibration, following guidance like Stein and colleagues' 2021 tutorial in Cell Reports Methods.
Ubiquitination stoichiometry: use Ub-AQUA or PSAQ standards to transform intensities into absolute amounts per site or linkage type; report per-protein normalization where appropriate (method foundations in Nature Methods).
Technical QC benchmarks for dual-PTM analysis
Identification and error rates: maintain ≤1% FDR at PSM/peptide levels; report site-level false localization rates around 5% with tool-specific probabilities (e.g., ptmRS thresholds). Independent FLR estimation and combined probability thresholds are discussed by Ramsbottom and colleagues in 2022 in Molecular & Cellular Proteomics.
Variability: target intra-batch median CV at or below 15% for well-controlled DIA-based workflows; use pooled references and bridge samples for inter-batch normalization.
Enrichment performance: document phospho enrichment yield and specificity; for diGly, report percentage of K-ε-GG-modified peptides in enriched fractions and note any cross-reactivity controls. Vendor documentation such as the CST K-ε-GG kit overview is useful for method parameters.
Validating crosstalk via kinetic modeling and PRM
Design perturbations that modulate priming kinases or pathways, then time-course samples to measure phospho-degron installation, diGly accumulation, and substrate half-life changes. Verify key sites with PRM; confirm localization and quantify dynamics relative to controls. Combine with proteasome or DUB inhibitors to parse degradation versus signaling roles.
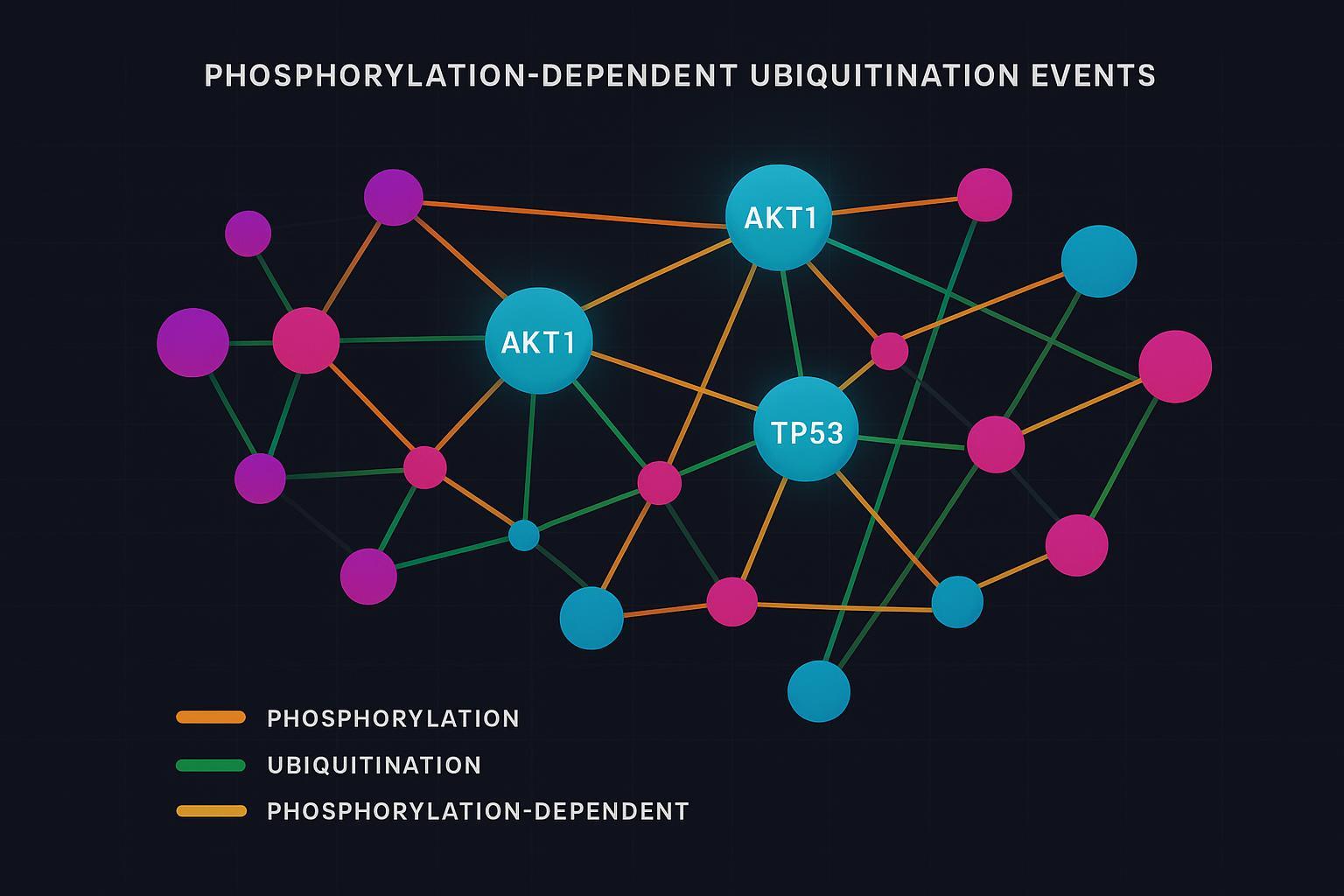 Figure 3: Systems-level map of phosphorylation–ubiquitination crosstalk across proteins.
Figure 3: Systems-level map of phosphorylation–ubiquitination crosstalk across proteins.
Bioinformatics integration and governance
Mapping the phospho–ubiquitin interaction map
DDA pipeline: MaxQuant or FragPipe with phospho and diGly variable modifications, 1% peptide FDR, site-localization probabilities exported per site. Merge site tables by protein and residue, then compute co-occurrence and motif enrichment using site-centered windows to reveal β-TrCP and FBXW7 consensus matches.
DIA pipeline: Spectronaut or DIA-NN, library-free where feasible. Export site-level probabilities for phospho and diGly, apply consistent probability thresholds, and use reference-channel or pooled-sample normalization. Visualize networks with Cytoscape and perform statistical tests for associations.
For a neutral overview of PTM proteomics scope and typical strategies, see the PTM proteomics hub.
NDA-compliant data governance and milestones
Adopt ALCOA+ principles for data integrity under NDA. Implement versioned SOPs, role-based access controls, audit trails, and standardized deliverables: raw data, search archives, localization tables, QC dashboards, and PRM reports. Define milestones upfront—pilot feasibility, method lock, cohort acquisition, PRM verification—and gate progression with QC sign-offs. Cross-reference cohort designs with repository deposition policies for public datasets when appropriate.
Case notes in proteostasis – Tau and AKT signaling
Dual-modification profiling in neurodegeneration
In Alzheimer's disease, phosphorylated tau epitopes (for example around S396 S404) often co-occur with ubiquitin signals within tangles, consistent with attempts at degradation and subsequent aggregation when clearance fails. Cohort-friendly DIA paired with diGly enrichment can track these signals alongside occupancy estimates and PRM for sentinel sites; see mechanistic and pathology-focused reviews such as Alquezar and colleagues' 2021 overview in Frontiers in Neurology.
Targeting E3 ligases via phospho-specific recruitment
Phospho-degrons in oncogenic substrates such as Myc or cyclin E recruit FBXW7, while β-catenin and IκB are degraded via β-TrCP. In pathway-centric studies, a time-course under kinase modulation can expose degron installation and subsequent ubiquitination—verifiable with PRM on degron-centered peptides; consensus and structural insights are synthesized in Science Advances and PNAS.
Future directions – proteogenomics and targeted degraders
Integrating global phosphorylation databases with ubiquitin maps
Large-scale phosphorylation resources can be combined with ubiquitin linkage and site maps to predict degron installation under specific kinase states. Motif-centric models and machine learning can prioritize candidate phospho-degrons for targeted validation.
From crosstalk to protein degraders
Crosstalk mapping informs degrader design by indicating when phosphorylation preconditions E3 recruitment. Degrader campaigns can incorporate PRM-ready assays for degron occupancy and ubiquitin buildup to confirm mechanism of action in cells.
Practical resources and neutral disclosure
Proteomics Methods Team, Creative Proteomics. We develop and implement quantitative PTM workflows focused on phosphorylation–ubiquitination crosstalk, sequential enrichment, and targeted PRM verification. Representative resources: the Creative Proteomics PTM hub. Disclosure: Creative Proteomics provided internal method resources referenced here. For research use only.
For a broad overview of PTM proteomics strategies and instruments in a single place, see the PTM proteomics hub by Creative Proteomics. Disclosure: Creative Proteomics is our product.
For study designs requiring high-throughput multiplexing, see the TMT-based proteomics overview.
For acquisition strategy trade-offs in phosphoproteomics, a neutral comparison of DIA vs DDA summarizes missingness and reproducibility considerations.
All resources above are for research use only.

Download

Our products and services are for research use only.
