
Protein kinases operate by transferring a phosphate group from ATP onto serine, threonine, or tyrosine residues on a protein polypeptide substrate, thereby directly influencing its activity and function. Radioactive studies have demonstrated that approximately 30% of proteins in eukaryotic cells undergo post-translational modifications via phosphorylation. This pivotal post-translational modification regulates a broad range of cellular activities, encompassing cell cycle progression, differentiation, metabolism, and neuronal communication. Furthermore, aberrations in phosphorylation events are correlated with numerous disease states.
The selection of methods for assessing phosphorylation may vary, contingent on several considerations, including the specific research question posed and the availability of specialized equipment or reagents. This paper will elucidate several frequently employed techniques for detecting protein phosphorylation, delineating the advantages and disadvantages inherent in each approach.
Select Service
Kinase activity assay
Protein kinase activity can be appraised via in vitro kinase assays, which involve mixing the isolated kinase with an unphosphorylated substrate and radioactively labelled ATP. The level of substrate phosphorylation is subsequently analyzed through a radioactive phosphate group. As a frequent component in numerous signaling transduction networks, protein kinases exert influence over multiple downstream biological responses. Thus, evaluating the activity of specific kinases can offer valuable insights into parallel pathways. In biological samples, kinase activity is generally assayed in vitro by co-incubating immunoprecipitated kinase with an exogenous substrate in the presence of ATP, and then measuring specific kinase-induced substrate phosphorylation using colorimetric, radioactive, or fluorescent detection.
Although this approach yields information about specific kinase behavior, assessing enzyme activity in cellular extracts only reveals part of the signaling pathway. Our understanding of protein modifications is still limited, and in vitro activity analyses do not reflect potential functions of endogenous phosphatases. Consequently, the direct detection of phosphorylated proteins may offer more detailed information about how cells respond to external stimuli, as identifying phosphorylated peptides can reveal the expression and functional status of proteins.
Phosphorylation-specific antibodies development
Conventional direct detection methods for protein phosphorylation involve the co-incubation of cells with radiolabelled 32P-phosphate to obtain cellular extracts. Subsequently, the extracts are separated via SDS-PAGE and exposed on films. This intricate process involves multiple incubations over a course of several hours and employs radioactive isotopes. An alternative traditional method is 2D gel electrophoresis, a technique predicated on the assumption that phosphorylation alters the migration rate and isoelectric points of proteins.
Given the complexities and time-consuming nature of the aforementioned methods, the development of phosphorylation-dependent antibodies has been widely welcomed. In 1981, researchers successfully generated the first documented phosphorylation antibody in rabbits, using benzoyl phosphate conjugated to Keyhole Limpet Hemocyanin (KLH) as an antigen. This antibody broadly recognized proteins containing a phospho-tyrosine residue. Ten years later, multiple phospho-state-specific antibodies were successfully developed by immunizing rabbits with synthetic phosphorylated peptides. These peptides represented the amino acid sequences surrounding target protein phosphorylation sites. The immunized serum was then applied to peptide-affinity columns to generate highly specific immuno-reagents.
The advent of phospho-specific antibodies not only improved traditional methodologies but also facilitated the development of novel immunoblotting techniques. However, successful detection in any technique using phospho-specific antibodies depends on the specificity and affinity between the antibody and target phosphorylated protein. Therefore, careful evaluation of their performance and applicability is required while selecting and using these antibodies.
SDS-PAGE
SDS-PAGE technology can effectively separate proteins, often cell or tissue lysates, in a sample by size. By applying a negative charge to all amino acids uniformly through buffer and SDS in the gel, proteins can migrate in an electric field based on their size not their unique amino acid composition or charge state. Proteins bands are clearly visible with Coomassie blue or silver staining methods. Significantly, when employing Coomassie blue or silver staining in SDS-PAGE, the technique also has the ability to detect phosphorylated proteins. phosphorylation results in slower migration of proteins compared to their unphosphorylated counterparts. A distinct limitation of SDS-PAGE lies in its inability to rule out migration changes in proteins caused by other perturbations such as glycosylation. Thus, when applying this technique, analysis of the phosphorylation status is primarily carried out on purified proteins.
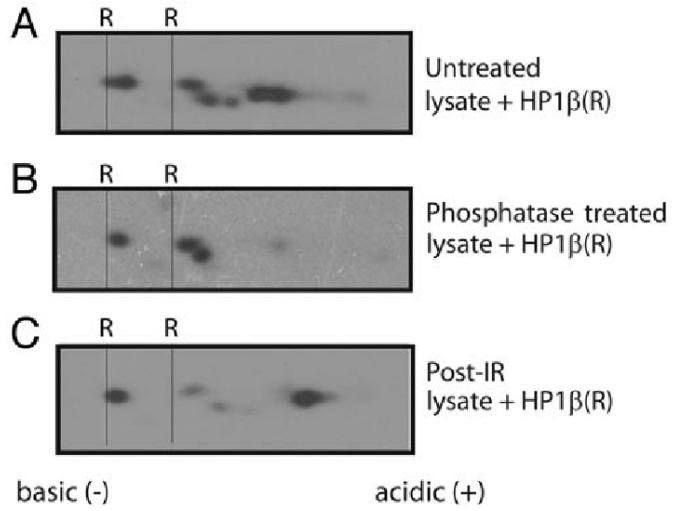 Phosphorylation of HP1β detected by 2D gel electrophoresis (Nabieh Ayoub 2019)
Phosphorylation of HP1β detected by 2D gel electrophoresis (Nabieh Ayoub 2019)
Mass spectrometry
A comprehensive assessment of protein phosphorylation (known as phosphoproteomics) within complex biological samples such as cell lysates is imperative for a nuanced understanding of phosphorylation-based signaling networks. This process involves large-scale analyses of phosphorylated proteins, including the identification of phosphorylated proteins and peptides as well as sequencing phosphorylated residues. Mass spectrometry (MS) techniques play a pivotal role in accomplishing these analyses.
In prevalent "bottom-up" MS approaches, proteins are digested into peptides, which are then ionized prior to the MS analysis. By comparing the compiled peptides with a protein database, based on their mass and fragmentation characteristics, each peptide is assigned to its corresponding protein. It should be noted that during this detection process, the signals of low-abundance proteins, such as phosphorylated proteins, could potentially be overshadowed by high-abundance proteins. Therefore, phosphorylated peptides are customarily enriched pre-MS analysis utilizing phosphorylation-specific antibodies or titanium dioxide.
Depending on the upstream sample preparation and matrix, a single MS scan could identify up to 5,000 proteins. Regardless of their phosphorylation states, MS can deliver semi-quantity (relative fold change) and quantity data (protein concentration). In contrast to methods like Western blotting and ELISA, MS boasts three distinct advantages. Firstly, MS is not antibody-dependent, hence it can identify new phosphorylation events, an analysis notion called "assumption-free" or "unbiased". Secondly, the dynamic range of Western blotting and ELISA is relatively narrow, typically spanning only 2-3 orders of magnitude, whereas MS can reach up to 5 orders of magnitude, allowing the detection of phosphorylated proteins at lower concentrations. Lastly, MS can easily differentiate among distinct protein isoforms.
While MS outperforms in terms of protein identification, challenges persist in the analysis of phosphorylated proteins. First, because phosphorylated peptides carry negative charges, and electrospray ionization MS operates in the positive mode, ionization is often inefficient, leading to generally weak signals for phosphorylated peptides. Second, signals from low-abundance target proteins can be difficult to observe in the high background of vast non-phosphorylated proteins. To overcome these limitations, researchers have developed various enrichment strategies, including Immobilized Metal Affinity Chromatography (IMAC), phosphorylation-specific antibody enrichment, and chemical modification techniques (such as β-elimination reactions of phosphoseryl and phosphothreonyl) and substituting phosphate groups with biotinylated moieties. These strategies help enhance the detection sensitivity and accuracy for phosphorylated proteins.
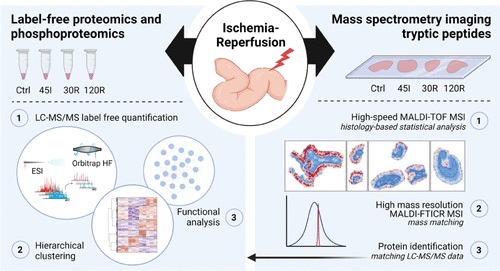 Phosphoproteome in the human intestine using liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis (J. Proteome Res. 2022)
Phosphoproteome in the human intestine using liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis (J. Proteome Res. 2022)
Through stringent R&D, our mass spectrometry-based phosphoproteomics platform integrates a variety of enrichment strategies, aiming to meet our clients' diverse experimental demands.
Select Service
Antibody array
Applying advanced sandwich antibody array technology, It is capable of conducting precise multiplex detection of phosphorylated proteins. In this process, the antibodies are meticulously immobilized onto surfaces such as glass, membranes, or magnetic beads. After the blocking steps are completed, we co-incubate the sample with the array. Subsequently, non-specific proteins are washed off and the array is co-incubated with detection antibodies for chemiluminescent or fluorescent analysis. The intensity of the signal is proportionate to the quantity of the antibody-bound protein. When we juxtapose the data from western blotting, ELISA, and the antibody array, all three techniques demonstrate analogous patterns in phosphorylation changes. Although the antibody array can offer quantitative data, the current market does not yet have quantifiable chip products for phosphorylated proteins. However, by adopting a readily addressable approach, we can easily discover or label different antibodies, thereby streamlining the protein identification process.
Western blot
Western blotting is currently the preferred method for evaluating the phosphorylation status of proteins, with many cell biology laboratories equipped to perform such experiments. This method separates biological samples through SDS-PAGE and subsequently transfers them to a membrane—typically, PVDF or nitrocellulose. Following this, target proteins are identified using antibodies specific for phosphorylation. The standard process for Western blot experiments eliminates the safety risks and waste disposal requirements incurred by the usage of radioactive isotopes.
Numerous phosphorylation-specific antibodies exhibit high sensitivity, enabling the effortless detection of phosphorylated proteins in common samples-- such as cell extracts in the range of 10-30 micrograms. Given the potential impact of handling or gel-loading errors on the measurement of phosphorylated protein levels, researchers frequently use one antibody to detect the total levels of a homologous protein (irrespective of its phosphorylation state). This benchmarking process facilitates the determination of the ratio of phosphorylated components to total components and serves as an internal control for loading.
Both chemiluminescent and colorimetric methods are extensively used, while molecular weight markers assist in providing information about the protein's molecular weight.
ELISA
As an indispensable tool, Enzyme-Linked Immunosorbent Assay (ELISA) has gradually become a crucial method for measuring protein phosphorylation. In comparative terms, ELISA supersedes Western blotting in its quantitative prowess, and showcases significant potential in investigating kinase activity and function. This microplate-based analytical method primarily relies on capture antibodies that are specific to the target protein, regardless of its phosphorylation state.
In this process, the target protein binds to an antibody-coated analysis plate; this protein could be a component of purified products or complex specimens such as the lysate of cells. Following this, a detection antibody pertaining to a particular phosphorylation site is introduced. These analyses generally employ colorimetric or fluorescent detection methods, and the intensity of the resulting signal is directly proportional to the concentration of the phosphorylated protein in the original sample.
Compared to traditional immunoblotting, phosphorylation-specific ELISA showcases substantial advantages. Firstly, quantification of results is readily achieved via calibrated standards. Secondly, the application of a sandwich-style dual antibody strategy significantly amplifies specificity. In addition, the high sensitivity of ELISA permits the use of fewer samples, thus enabling the detection of low abundance proteins. Finally, the microplate format materially enhances analytical throughput, rendering it more advantageous than traditional Western blotting.
It is noteworthy that ELISA is typically used for the indirect determination of kinase activity. Nevertheless, another version of ELISA technology employing immobilized capture antibodies, substrate, and detection methods for phosphorylated substrates provides a more direct means for measuring kinase activity.
In-Cell ELISA
"In-Cell ELISA" In what has been a widespread application for hypothesis testing and drug screening, biological biochemical kinase assays such as traditional sandwich Enzyme-Linked Immunosorbent Assays (ELISA) have played an immense role. However, these assays might not faithfully mirror the intricate environments within cells. Therefore, studying protein phosphorylation processes inside intact cells may provide a more authentic reflection of the state of specific signaling pathway networks.
Recently, several immunological assays have been developed that are designed for intact cells, capable of quantifying intracellular protein phosphorylation levels. In these methods, stimulation, fixation, and blocking processes are performed within the same well. Subsequently, using the specificity of antibodies against phosphorylation sites along with either a fluorescent or colorimetric detection system, protein phosphorylation states are evaluated.
Notably, these innovative methods are equipped with the capacity to concurrently measure phosphorylated proteins and total proteins within the same well. This design permits the use of a secondary protein signal for the normalization of the target protein signal, thereby compensating inter-well variances and ensuring accurate assessment of phosphorylated protein levels. This matches traditional immunoblotting techniques, where antibodies specific for phosphorylated proteins and total protein are utilized.
As these novel analysis methods eliminate the need to prepare cell lysates, they are better suited for high-throughput analysis. By reducing operation steps and sample processing time, they equip researchers with more robust, accurate tools for protein phosphorylation studies.
Flow Cytometry and ICC/IHC
The conventional intracellular flow cytometry and immunocytochemistry/immunohistochemistry (ICC/IHC) play pivotal roles in the detection of phosphorylation events. Flow cytometry exploits lasers to excite fluorescent dyes for antibody detection. When evaluating multiple proteins within a single cell, the judicious selection of filter combinations and fluorescent dyes is crucial to ensure their spectra do not overlap. The substantial advantage of flow cytometry lies in its capability to conduct rapid and quantitative single-cell analyses. By identifying cell surface markers, this technique can detect proteins in distinct cell types within heterogeneous populations without necessitating physical separation. This methodology is particularly applicable to the analyses of rare cell populations and effectively circumvents issues arising from cell loss or protein expression alterations during cell sorting processes.
Meanwhile, ICC generally refers to the microscopic examination of proteins within cultured cells, while IHC is intended for proteins within intact tissue sections. These techniques, akin to flow cytometry, can assess multiple proteins within cells or tissues, albeit with special attention required to avoid overlapping fluorescent spectra or colors. Both fluorescence and chromogenic detection techniques are broadly utilized in such applications. Compared to other means of monitoring phosphorylation, ICC is typically the preferred method for determining intracellular localization. Whether it's flow cytometry or ICC/IHC, the use of highly affinity-specific antibodies is required, together with the implementation of blocking steps, controls, and antibody titration measures, to obliterate potential interferences stemming from non-specific binding.
The use of flow cytometry and ICC for detecting phosphorylated proteins is predicated on the prerequisite that the protein remains stable, and the antibody can approach its target. Cells typically need to be stimulated and fixed via formaldehyde or paraformaldehyde to stabilize and cross-link phosphorylated proteins for subsequent analyses. Once fixed, cells also need to undergo a permeabilization process to allow access to phosphorylation-specific antibodies inside the cells. Different permeabilization techniques are required for different subcellular localizations. Mild detergents are suitable for cytoplasmic protein detection, whereas alcohol or other permeabilization agents might be necessary for nuclear protein detection. Alcohol permeabilization not only enhances the detection efficiency of phosphorylation-specific antibodies towards phosphorylated proteins but also offers unique advantages due to its denaturing properties.
Multiple Parallel Analysis through Mass Spectrometry
Mass spectrometry techniques, such as Collision-Induced Dissociation (CID) and Electron Transfer Dissociation (ETD), offer expansive parallel analytic capabilities in contemporary studies of peptide sequencing and post-translational modifications, particularly phosphorylation. Despite these approaches' high-depth analysis, comprehensive phosphorylation analysis strategies might not always be imperative when focused on specific pathway investigations. Consequently, a myriad of novel methods have been developed for simultaneous assessment of the phosphorylation state of multiple analytes.
These techniques primarily rely on the application of phospho-specific antibodies, encompassing detection platforms based on microplates, magnetic beads, and membranes. A considerable advantage that these newly-emerged methods possess is their substantial amplification of throughput capacity, thus curtailing the necessity for multiple Western blot or traditional enzyme-linked immunosorbent assay (ELISA) analyses. Furthermore, these methodologies can provide an abundance of data, even with minimal sample volumes.
Notably, however, proteomic analytic techniques generally exhibit lower sensitivity compared to their conventional counterparts, predominantly due to potential antibody cross-reactivity. Therefore, consideration of these methods' limitations is required, in conjunction with thoughtful selection and application relative to specific research objectives and conditions.
.jpg)
Download


Our products and services are for research use only.
