
Introduction of Post-Translational Modifications
Proteins, as the executants of biological functions, manage biological activities and metabolic pathways not solely through the modulation of their own expression levels. Instead, they drive fundamental cellular functions through rapid and reversible modifications. These encompass environmental change sensing, cellular communication, and even the destiny of cell life and death. To unravel the mysteries surrounding dynamic post-translational modifications (PTMs) of proteins and their complexes, high-throughput mass spectrometry data acquisition technologies have become powerful tools.
Post-translational modifications (PTMs) refer to the covalent breakage or formation on a protein backbone or the side chain of an amino acid. Notably, PTMs have a distinct advantage in eukaryotes compared to prokaryotes; approximately 5% of the eukaryotic genome encodes enzymes involved in protein modifications. The known PTMs exceed over 400 types to date, each named according to the functional groups transferred onto the amino acid residues, including phosphorylation, glycosylation, methylation, and ubiquitination. Common protein PTMs consist of phosphorylation, glycosylation, methylation, ubiquitination, acetylation, and oxidation. PTMs can be classified into two types of modifications: enzymatically catalyzed modifications (like phosphorylation, glycosylation) and covalent cleavage (like glycosylation, oxidation). In enzyme-mediated PTMs, electrophilic groups covalently bond to nucleophilic side-chain residues; non-enzymatic modifications occur solely through the cleavage of peptide backbones. Among the known 22 proteinogenic amino acids, only leucine, isoleucine, valine, alanine, and phenylalanine are not modified in vivo; all other amino acids can undergo a variety of different modifications, such as methylation, acetylation, or ubiquitination of lysine.
Post-Translational Modifications Analysis Workflow
PTMs significantly expand the functional diversity of proteins, having a role in numerous physiological and pathological biological processes. Therefore, identifying and elucidating PTMs have profound implications for disease prevention and treatment. Mass spectrometry-based methods are commonly used for PTMs research, involving several steps. Given that most PTMs exhibit transient, unstable, and low chemical quantities (at the femtomole level), the initial step before mass spectrometry scanning is enrichment, which can be conducted through chemical methods or selective enrichment for modified residues using antibodies. Subsequent steps involve mass spectrometry scanning of the peptides resulting from protein hydrolysis. In MS mode, the parent ion mass-to-charge ratio of the modified peptide can be detected and compared to the designed peptide theoretical mass-to-charge ratio to determine the occurrence and quantity of modifications. MS/MS fragment ions help sequence the modified peptide, thereby identifying and locating the position of the mass shift. However, determining a mass shift might pose challenges under certain circumstances. For instance, the mass shift of trimethylation and acetylation is similar, necessitating the use of highly sensitive and accurate MS equipment for differentiation.
Select Service
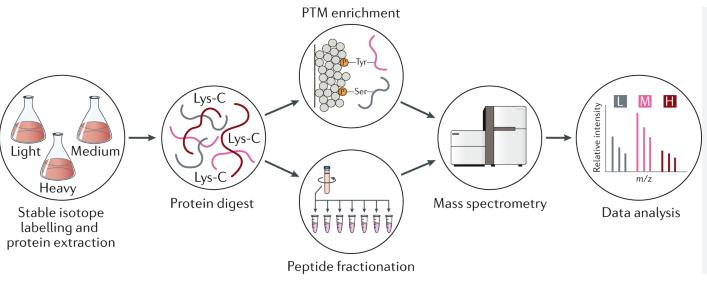 Protein post-translational modifications workflow (Boris Macek et al,. 2019)
Protein post-translational modifications workflow (Boris Macek et al,. 2019)
Protein Phosphorylation
Protein phosphorylation, a widely studied reversible protein modification process, occurs via the covalent bond between the phosphate group and serine, threonine, or tyrosine. This modification is widely understood to impact protein synthesis, activity, stability, and regulate intercellular/intracellular signal transmission, gene expression, cell survival, and apoptosis. Uncontrolled phosphorylation events might lead to the development of various diseases, such as cancer, so the identification and characterization of phosphorylated proteins can help clarify related pathological mechanisms.
Protein phosphorylation and dephosphorylation processes are catalyzed by kinases and phosphatases, respectively. Kinases catalyze the formation of phosphate ester bonds, transferring the phosphate group from adenosine triphosphate or guanosine triphosphate to serine, threonine, or tyrosine. This bond can then be hydrolyzed by phosphatases, releasing the phosphate group. Given that the kinetics of phosphatases are much faster than those of kinases, the deactivation of phosphatases to necessary in proteomic phosphorylation studies. To date, over 500 human kinases and 150 phosphatases have been discovered, confirming the pivotal part this modification plays in cellular and biological events.
In the field of phosphopeptide enrichment, the foremost strategies include titanium dioxide affinity chromatography and immobilized metal affinity chromatography (IMAC). To identify more phosphorylated peptides, it's common to fractionate the sample using chromatographic techniques, such as strong cation exchange chromatography or high pH reverse-phase chromatography, to reduce complexity before enrichment. Additionally, combining two or more phosphopeptide enrichment strategies can further increase the number of identified phosphorylation sites—like the combination of titanium dioxide affinity chromatography and IMAC, known as TIMAC. For peptide segments with phosphoryl modifications, there's also relevant research on antibody enrichment methods with a particular emphasis on tyrosine phosphorylated modification segments. Due to the close connection between tyrosine phosphorylation and the development of cancer, exploration in this area carries significant weight.
Select Service
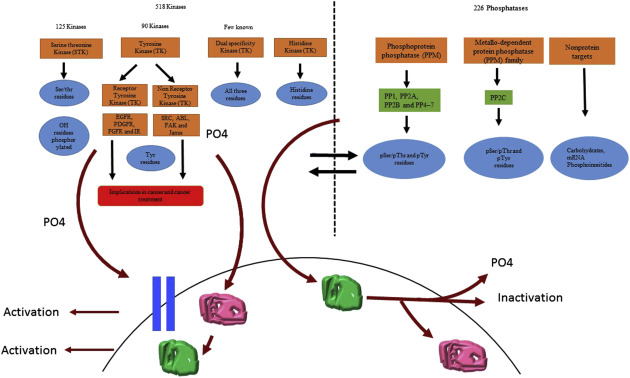 Protein phosphorylation Modification (Sabita N. Saldanha et al,. 2018)
Protein phosphorylation Modification (Sabita N. Saldanha et al,. 2018)
Protein Ubiquitination
Ubiquitination is among the most widely researched primary regulatory modifications. This enzymatic process involves one or more ubiquitin molecules (Ubiquitin, Ub) covalently binding to the lysine residues of substrate protein molecules under the catalytic action of enzymes E1, E2, and E3. Ubiquitin is a highly conserved 76-amino acid polypeptide in eukaryotic organisms, including seven lysine residues and one N-terminal (M1, K6, K11, K29, K33, K48, and K63). These residues are ubiquitinated, prompting the assembly of polyubiquitin chains. These chains possess unique structures and confer different biological functions to organisms. For instance, K48 chains are associated with proteasomal degradation, while K63 chains are involved in cell signal transduction and lysosomal degradation. Given that ubiquitin chains can be comprised of a single type, mixed, or branched, numerous topological structures of ubiquitin chains exist, forming a complex signaling network.
The process of ubiquitin modification involves the collaborative action of ubiquitin-activating enzyme E1, ubiquitin-conjugating enzyme E2, and ubiquitin ligase E3. E1 activates ubiquitin in an ATP-dependent manner, transferring ubiquitin to the active cysteine site of E1 and releasing adenosine monophosphate. Subsequently, ubiquitin binds with the active cysteine site of E2, enabling E3 to link ubiquitin to specific lysine residues on proteins via thioesterification. Ubiquitination does not rely on sequence recognition motifs but is dependent on the availability of lysine residues, and the specificity of E3 enzymes or E2/E3 enzymes.
Mass spectrometry analysis of ubiquitinated peptides is achieved by proteolytic digestion of the protein sample. As the two C-terminal glycine residues of ubiquitin bind to lysine residues, and trypsin cleaves at the C-terminal of lysine, it produces a characteristic di-glycine (diGly) signal. Consequently, compared to unmodified peptides, the mass of ubiquitinated peptides increases by 114.0429 Da, aiding in the prediction of ubiquitination sites. Identification and quantification of ubiquitin modifications require enrichment of the sample, commonly realized using antibodies.
Select Service
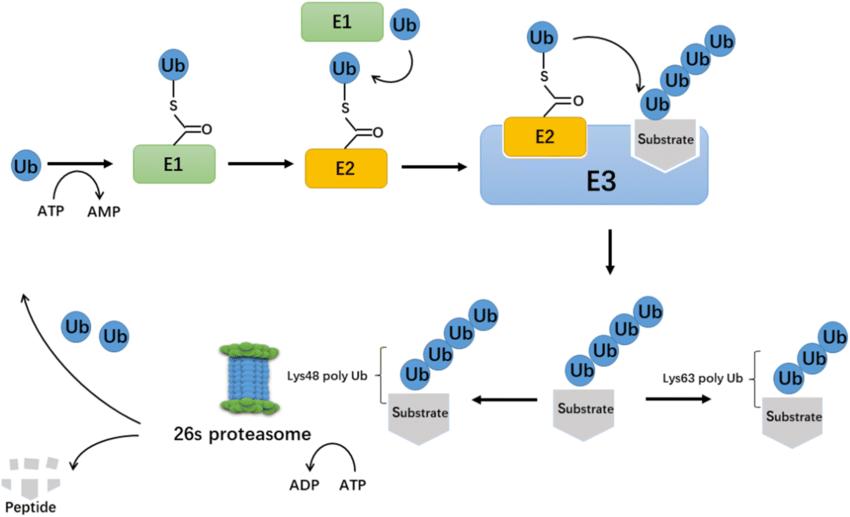 The process of ubiquitination. (Xiaofeng Gong et al,. 2020)
The process of ubiquitination. (Xiaofeng Gong et al,. 2020)
Protein Acetylation
Initially discovered in histones, acetylation has since been identified in over 80 transcription factors and other nuclear regulatory biomolecules. Protein acetylation has been confirmed to participate in numerous cellular processes including transcriptional and metabolic regulation. Research demonstrates that the majority of enzymes involved in glycolysis, gluconeogenesis, tricarboxylic acid cycle, urea cycle, fatty acid metabolism, and glycogen metabolism are acetylated. Histone acetylation affects gene transcription, whereas histones without acetylation inhibit transcription. Histone acetyltransferases (HATs) transfer an acetyl group from acetyl coenzyme A to the amino group of a histone's terminal lysine, forming N-acetyllysine, while histone deacetylases (HDACs) perform the opposite process. As a result, excessive protein acetylation could potentially lead to obesity, cancer, and neurodegenerative disorders, rendering HATs and HDACs as potential drug targets for these diseases.
In the mass spectrometric analysis of acetylation, strategies similar to those previously discussed for protein post-translational modifications (PTMs) are employed. Initially, proteins are broken down into peptides using trypsin. The standard enrichment method involves immunopurification using an acetyllysine antibody, followed by analysis with liquid chromatography-tandem mass spectrometry (LC-MS/MS). Acetylated peptides exhibit a 42.0367 Da mass shift. The acetylation modification stymies trypsin cleavage by neutralizing lysine charges, thereby serving as evidence of lysine modification.
Select Service
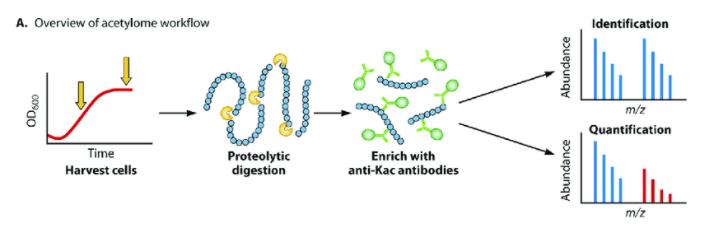 Typical acetylome workflow. Jackson (Luu et al,. 2021)
Typical acetylome workflow. Jackson (Luu et al,. 2021)
Protein Methylation
Protein methylation represents a form of biological modification, whereby a methyl group is transferred from S-adenosylmethionine (SAM) to the side chain of specific methyl acceptors, such as lysine, arginine, histidine, aspartamide, and glutamamide. Among diverse methylation modalities, lysine and arginine methylation are particularly prevalent. Notably, lysine methylation is a reversible chemical modification process.
Three distinct forms of lysine methylation exist, mediated by Protein Lysine Methyltransferases (PKMTs), namely monomethylation (Kme1), dimethylation (Kme2), and trimethylation (Kme3). The various methylation states have profound implications for biological processes including heterochromatin formation, X-chromosome inactivation, and transcriptional silencing or activation.
On the other hand, lysine demethylases, based on the differences in catalyzed reaction types, are principally divided into two families: LSD and JmjC-domain-containing demethylases. The human genome encodes two LSD enzymes (LSD1 and LSD2), which can only demethylate mono- and dimethylated lysine. The demethylation of trimethylated lysine, by contrast, relies on demethylases harboring a JmjC domain.
Select Service
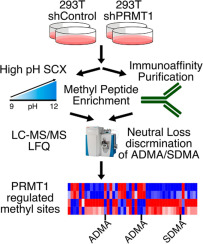 Protein methylation analysis workflow (Nicolas G. Hartel et al,. 2019)
Protein methylation analysis workflow (Nicolas G. Hartel et al,. 2019)
Protein SUMOylation Modifications
SUMOylation refers to the process of covalently attaching a highly conserved small molecule protein family, known as Ub-like modifiers (SUMOs), to specific lysine residues in a protein. Five SUMO proteins (SUMO1-5) have been discovered in eukaryotic organisms. SUMO2 and SUMO3 share a sequence similarity of up to 97%, and they are typically collectively referred to as SUMO2/3. SUMO1, SUMO2, and SUMO3 are widely expressed in all cells and organs, while SUMO4 is specifically expressed in certain organs such as the kidneys, lymph nodes, and spleen. SUMO5 is mainly expressed in the lungs and spleen.
The SUMOylation process resembles the Ub modification, involving the participation of SUMO activating enzyme (E1), SUMO conjugating enzyme (E2), and SUMO ligase (E3). Reported E3 ligases include members of the PIAS protein family, hPC2, and RanBP2. SUMOylation is a dynamic and reversible process, with deSUMOylation primarily regulated by SENPs.
SUMOylation is closely associated with protein expression, localization, stability, and activity regulation, participating in various cellular processes such as protein-protein interactions (PPI), intracellular localization, DNA repair, nucleocytoplasmic transport, transcription factor (TF) activation, cell apoptosis, the cell cycle, and gene transcription, among others. The mechanisms of SUMOylation impact can be classified into two categories: one directly influences protein function through covalent modification of proteins, whereas the other indirectly regulates protein biological function through the non-covalent binding of target protein with SUMO interaction motif (SIM).
Although SUMO and Ub are similar in structure and process of occurrence, their effects often oppose each other. Ubiquitinated proteins are usually directed to the proteasomes for degradation, whereas SUMOylated proteins are generally more stable and less prone to degradation. The K21 in IκBα is an illustrative example, where SUMOylation and ubiquitination mutually antagonize each other.
Select Service
Proteins Glycosylation
Glycosylation is a process where glycans are enzymatically added to proteins via glycosyltransferase, transferring the glycan from a donor molecule to the protein. These reactions predominantly take place in the endoplasmic reticulum (ER) and the Golgi apparatus. The action of glycosyltransferase constructs a variety of complex glycans, prominently found on the cell surface and secretory proteins. Glycosylation reactions that occur in the nucleus and cytoplasm tend to result in simpler structures, exhibiting highly dynamic features.
There exist four types of linkages between glycans and proteins.
N-Glycosylation primarily happens at asparagine (Asn) residues in the protein structure. A sugar complex centered on GlcNAc2Man3 links to the Asn side chain's nitrogen atom through GlcNAc, recruiting relevant enzymes to remove or add monosaccharides. After ER processing, the precursor's polysugar is moved to the cis-Golgi, followed by modifications carried out by specific mannosidases. It then enters the Golgi for further processing and maturation.
O-Glycosylation primarily occurs on amino acids such as serine (Ser) and threonine (Thr) that bear functional hydroxyl groups on the side chains, with the relevant monosaccharides being primarily GalNAc and GlcNAc. GalNAc type O-glycosylation is predominantly present in secreted glycoproteins and those found outside of cells; this type of glycosylation initiates in the Golgi apparatus.
C-Glycosylation, a less common type of protein glycosylation, chiefly operates on proteins found in the ER. It plays a critical role in protein folding, sorting, and secretion processes.
Glypiation, a unique type of glycosylation, employs glycosylphosphatidylinositol (GPI) to position proteins onto the cell membrane. This process is of paramount importance in cellular signal transmission, cell adhesion, and immunorecognition.
Select Service
Proteomics Analysis of Glypiation
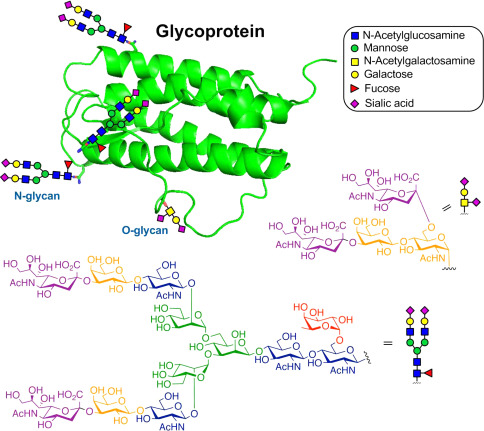 The structure of a typical glycoprotein (Yaohao Li et al,. 2019)
The structure of a typical glycoprotein (Yaohao Li et al,. 2019)
Proteins Citrullination
Protein citrullination is the irreversible process of converting arginine residues in proteins to citrulline residues under the action of peptidylarginine deiminases (PADs). This can affect hydrogen bond formation, protein folding, hydrophobicity, and protein-protein interactions, ultimately leading to protein denaturation. The most common substrates for PAD-mediated citrullination include keratin, fibrinogen, vimentin, myosin, histones, collagen, and myelin basic protein. Notably, free arginine cannot be citrullinated by PADs.
Peptidyl Arginine Deiminases, PDAs, are comprised of five isozymes (PDA1-4 and PDA6), each exhibiting unique tissue-specificity. Among those, PDAs 1-4 have catalytic activity, while PDA6 becomes catalytically inactive due to a mutation at its active site. Despite shared sequence homology, PDAs are distinguished by their distinctive tissue distribution.
PDA1 and PDA3 chiefly reside in hair follicles and the epidermis, with PDA1 also making its presence in the uterus. PDA2 is the most wide-spread PDA in the human body. Its two primary substrates are myelin basic protein and Glial Fibrillary Acidic Protein (GFAP), both of which are found in the central nervous system.
PDA4 is present in neutrophils, macrophages, mammary cells, and tumor cells majorly catalyzing histone citrullination, disrupting DNA and hence leading to cell apoptosis. Notably, PDA4 plays a role in histone citrullination in neutrophils. Upon activation, PDA4 translocates to the nucleus of neutrophils, and forms neutrophil extracellular traps (NETs) to ensnare bacteria and other pathogens. Interestingly, PDA6 is found in early embryos, oocytes, and ovaries.
Select Service
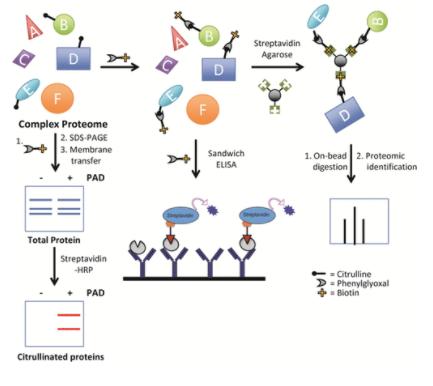 Protein citrullination analysis (Daniel M. Lewallen et al,. 2015)
Protein citrullination analysis (Daniel M. Lewallen et al,. 2015)
ADP-ribosylation
ADP-ribosylation refers to the process operated by ADP-ribosyltransferases that transfers ADP-ribose from NAD to a target protein at its ADP-ribose binding domain. This process plays a pivotal role in the structural and functional regulation of numerous proteins, among them being nuclear protein topoisomerase I, DNA ligase II, endonuclease, histones H1, H2B, and H4, DNA polymerase, as well as cytoplasmic proteins like adenylate cyclase and elongation factor eEF-2. ADP-ribosylation is significantly involved in an array of physiological and pathological processes, such as signal transduction, protein transportation, transcription, DNA damage repair, cell cycle regulation, as well as apoptosis and necrosis.
Select Service
Protein Benzoylation
Lysine benzoylation (Kbz), the first discovered aromatic fatty acid modification, primarily acts on the N-terminal tails of histones. Sodium benzoate (SB) can be transformed into benzoyl-CoA in mammalian cells, serving as a precursor for Kbz and a key intermediary in the breakdown of numerous aromatic growth substrates in bacteria and gut microbiota. This type of modification is physiologically distinct from histone acetylation. Kbz predominantly operates at gene promoters, regulating pathways including glycerophospholipid metabolism, ovarian steroidogenesis, and phospholipase hydrolysis signaling. An excessive intake of SB may lead to elevated Kbz levels, thereby potentially escalating the risk of disorders such as motor coordination impairment and Attention Deficit Hyperactivity Disorder (ADHD).
Select Service
Protein Neddylation
Neddylation is a chemical modification process that operates on proteins. The process couples the Ub-like protein NEDD8 to a target protein, thereby influencing various facets of protein function, including protein conformation, stability, subcellular localization, DNA affinity, and substrate binding. Similar to ubiquitination, Neddylation exerts impact on multiple stages such as proliferation, apoptosis, and migration in cancer cells.
NEDD8 is a highly conserved protein that possesses high sequence and structural similarities with Ubiquitin (Ub). The NEDD8 activating enzyme E1, composed of an APPBP1/UBA3 heterodimer, catalyzes the formation of high-energy thioester bond between the C-terminal glycine of the NEDD8 molecule and the active cysteine site of UBA3 through its ATP-dependent catalytic subunit, thereby activating the NEDD8 molecule. The NEDD8-charged NEDD8 activating enzyme (NAE) can transfer to the E2 conjugating enzyme UBC12/UBE2M or UBE2F via a transthiolation reaction. Eventually, substrate-specific E3 ligase covalently transfers NEDD8 from E2 to the lysine residue within the target protein. It's noteworthy that Neddylation is a reversible process where NEDD8 can be dissociated from the substrate protein under the actions of deneddylase.
Select Service
SCFA Modification
Short-chain fatty acids (SCFAs) are products of food digestion and dietary fibre fermentation in the gut, characterized by a carbon atom count of no more than six. SCFAs play extensive roles in signal transduction, cellular metabolism, and immunity. These molecules could be converted into acyl-coenzyme A, serving as donors for lysine acylation in proteins, and modifying the lysine side chains via various Histone Acetyltransferases (HATs).
In addition to acetylation (the most extensively studied type of SCFA modification to date), a range of other SCFA-derived modifications have been identified. These include propionylation (Kpr), butyrylation, 2-hydroxyisobutyrylation, succinylation, isobutyrylation, malonylation, glutarylation, crotonylation, β-hydroxybutyrylation, and lactylation (Kla). Concurrently, acylated proteins can undergo deacylation by Histone Deacetylases (HDACs) and sirtuins.
LCFAs Modification
Long-chain fatty acids (LCFAs) refer to straight-chain fatty acids whose molecular structure contains 12 or more carbon atoms, such as myristic acid, linoleic acid, and palmitic acid. Similar to short-chain fatty acids (SCFAs), LCFAs can also link with the N-terminus and the side chain of amino acids through enzymatic action. Within LCFAs, palmitoylation and myristoylation are the most common forms of modifications. Palmitoylation plays a key role in regulating the translocation, localization, and stability of proteins, while myristoylation holds significant importance in innate immunity, signal transduction, and cancer progression.
Summary
PTMs of proteins play a pivotal role in dictating cellular phenotypes and biological processes. The maintenance of the equilibrium state of protein modifications holds significant implications for human health. Aberrations in PTMs could potentially lead to alterations in protein characteristics and loss of function, which are closely tied to the onset and progression of numerous diseases. To our latest knowledge, over 600 types of PTMs have been identified and recognized. This overview aims to detail some of the more popular types of protein modifications to enhance the understanding of PTMs, and to foster deeper exploration into the interplay between target proteins and inducing models in the course of research.
.jpg)

Related Articals
Our products and services are for research use only.
