
Methods for the Identification of Protein Modification Sites
The identification of protein modification sites is crucial for understanding the functional regulation of proteins. Protein modifications, such as phosphorylation, ubiquitination, acetylation, and methylation, play critical roles in cellular processes. These modifications can alter protein activity, localization, interaction, and stability. Various methodologies are employed to identify these modification sites, each with distinct advantages and applications.
In the investigation of protein post-translational modifications (PTMs), initial efforts typically encompass two primary scenarios: the comprehensive screening of known or previously reported modification sites, and the exhaustive de novo identification of novel sites. These processes aim to delineate candidate sites that are closely associated with the specific research focus.
Based on the diverse objectives and specific requirements of PTM research, strategies for identifying protein modification sites can be broadly categorized into two main approaches: mass spectrometry-based identification and antibody microarray screening analysis. Given Creative Proteomics' expertise in high-throughput identification technologies centered around mass spectrometry, this paper will focus on elucidating the fundamental principles and methodologies of mass spectrometry for the identification of protein modifications.
Mass Spectrometry Identification Method
Principle and Process of Mass Spectrometry Identification
Over the past two decades, MS has emerged as an essential analytical technique for the determination of protein modification types and sites. For the identification of modified peptides and PTMs, peptides are initially separated using reversed-phase chromatography. This is followed by the collection of fractions and subsequent mass spectrometric analysis (LC/MS). To accurately ascertain the nature of peptide modifications, tandem mass spectrometry (MS/MS) is frequently employed. In the initial MS step, peptide ions are subjected to fragmentation by collision with an inert gas, resulting in additional fragmentation products. These peptide fragments are subsequently analyzed in a secondary MS step. During this process, certain modified peptides may remain intact, yielding fragmentation patterns akin to those of unmodified peptides, while other peptides may undergo extensive fragmentation, thereby providing enhanced information on the nature and localization of the modified amino acids.
Select Service
Additional Applications of Mass Spectrometry
Broad Dataset Characterization
MS extends beyond the analysis of individual protein modifications to encompass the comprehensive characterization of broad datasets, enabling the exploration of specific protein subsets, often referred to as "sub-proteomes." This capability is particularly valuable in the study of PTMs, such as phosphorylation. For instance, phosphoproteomics—a sub-discipline focused on phosphorylated proteins—utilizes MS to map phosphorylation sites across numerous proteins within a biological sample. This mapping provides a detailed overview of cellular signaling pathways, their regulation, and their alteration under different physiological and pathological conditions.
Analysis of Signaling Networks
The application of mass spectrometry in PTMs proteomics has proven instrumental in deciphering complex signaling networks. These networks, often deregulated in diseases such as cancer, can be studied comprehensively to understand the activation states of various pathways. For example, Olsen et al. (2010) utilized MS-based phosphoproteomics to perform a global analysis of phosphorylation events in HeLa cells. This study identified over 6,000 phosphorylation sites, offering insights into the regulation of key signaling pathways, including those involved in cell cycle control and apoptosis. The ability to quantify phosphorylation changes provides a dynamic view of signaling network alterations in response to external stimuli or disease progression.
Sub-Proteome Identification
The identification of specific sub-proteomes, such as the phosphoproteome, via MS enables researchers to concentrate on subsets of proteins that share functional properties or regulatory mechanisms. This methodological approach has proven particularly advantageous in cancer research, where the comprehensive characterization of the phosphoproteome can uncover aberrant signaling pathways that drive tumorigenesis. By identifying differentially phosphorylated proteins, researchers can isolate potential biomarkers for diagnosis, prognosis, and therapeutic intervention.
For instance, Mann et al. (2002) employed MS to profile the phosphoproteome of colorectal cancer cells. Their findings emphasized critical proteins exhibiting altered phosphorylation patterns, which could serve as biomarkers for the disease. This study exemplifies how mass spectrometry not only facilitates the identification of post-translational modifications but also enhances the understanding of disease mechanisms at the molecular level.
Limitations of Mass Spectrometry in Identification
Despite its strengths, MS faces certain limitations. Not all peptide fragments can be detected; some are clearly visible, while others remain undetectable, which poses a significant challenge for analyzing complex mixtures. Additionally, due to the low abundance and ionization efficiency of modified peptides, their signals can be suppressed by those of unmodified peptides. Therefore, prior to MS-based identification of protein modifications, enrichment techniques are often required to enhance signal-to-noise ratios. This typically involves affinity chromatography before mass spectrometric analysis.
Enrichment Methods
Different types of modifications necessitate distinct enrichment methods, as outlined in Table 1.
| PTM Type | Enrichment Method |
|---|---|
| Phosphorylation | Metal Ion Affinity Chromatography (IMAC, TiO2), Motif Antibody Enrichment |
| Acetylation | Motif Antibody Enrichment |
| Methylation | Motif Antibody Enrichment |
| Ubiquitination and Ubiquitin-like Modifications | Motif Antibody Enrichment, Tag-specific antibodies for tagged ubiquitin or ubiquitin-like proteins |
Definition and Distinction of Motif Antibodies vs. Modification Site-Specific Antibodies
Motif Antibodies
A motif, in the context of biological macromolecules such as DNA and proteins, refers to a conserved sequence or arrangement of nucleotides or amino acids. For protein modification sites, a motif typically denotes the sequence pattern surrounding an amino acid residue that undergoes a specific modification. For example, the antibody Phospho-PKASubstrate (RRXS*/T*) (100G7E) Rabbit mAb #9624 targets the PKA (Protein Kinase A) phosphorylation site within the substrate sequence (R)(R)X(S/T). This sequence is characterized by the presence of arginine (R) at positions -2 and -3 relative to the phosphorylated serine/threonine (S/T), while X represents any amino acid at position -1. Antibodies that recognize these sequence motifs, encompassing various permutations of amino acids surrounding modification sites, are known as motif antibodies.
Modification Site-Specific Antibodies
In contrast to motif antibodies, which recognize a broad array of peptide sequences containing modifications, site-specific antibodies are designed to precisely identify a particular modification at a specific site on a protein. Site-specific antibodies are characterized by their specificity to a single modification site, with recognition dependent on the precise sequence surrounding the modification.
Summary of Differences
Scope of Recognition: Motif antibodies are capable of identifying a wide range of peptide sequences and modifications due to their recognition of specific sequence motifs surrounding modification sites. Conversely, site-specific antibodies are tailored to recognize modifications at precise locations on a protein, with a defined amino acid sequence context.
Application: Motif antibodies are useful for detecting multiple potential modifications across various proteins, while site-specific antibodies provide targeted analysis of individual modification sites.
Antibody Design: Motif antibodies are designed to bind to common patterns of amino acid sequences associated with modifications, whereas site-specific antibodies are developed to bind to unique sequences that include a specific modification.
Understanding the distinction between motif antibodies and site-specific antibodies is crucial for selecting appropriate tools for studying protein modifications and their biological implications.
Mass Spectrometry: High-Throughput Assessment of Known Modification Sites
Mass spectrometry is a powerful analytical technique utilized for high-throughput assessment of both unknown and known protein modification sites. While it is commonly employed to identify novel post-translational modifications and characterize unknown modification sites, it is equally effective for the high-throughput evaluation of known modification sites.
High-Throughput Evaluation of Known Modification Sites
Mass spectrometry enables the comprehensive analysis of known modification sites by providing detailed insights into their abundance, distribution, and dynamics within a sample. This capability is particularly advantageous for:
Quantitative Analysis: Mass spectrometry can quantify the extent of modification at specific sites, allowing for the assessment of modification levels under various experimental conditions or treatment regimens.
Reproducibility and Precision: The technique offers high reproducibility and precision, ensuring accurate measurement of modification sites across multiple samples and experimental runs.
Contextual Analysis: By integrating mass spectrometric data with additional proteomic information, researchers can elucidate the context of known modifications within the broader protein structure and function.
Comparative Studies: Mass spectrometry facilitates comparative studies by enabling the simultaneous evaluation of known modification sites across different biological samples, conditions, or time points.
Overall, mass spectrometry provides a robust and versatile platform for the high-throughput analysis of known protein modification sites, complementing its utility in discovering and characterizing novel modifications.
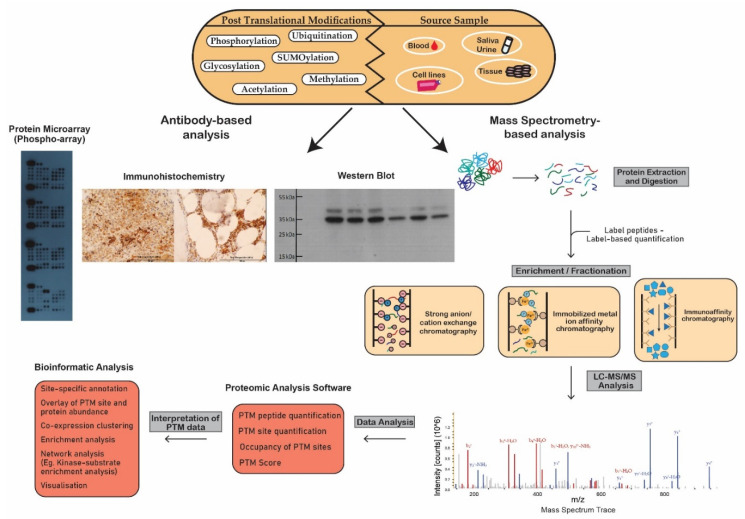 Figure 1 Schematic workflow illustrating analytical techniques used in the analysis of post-translational modifications.
Figure 1 Schematic workflow illustrating analytical techniques used in the analysis of post-translational modifications.
Antibody Microarray-Based Screening Analysis
Antibody microarray-based screening is a powerful technique employed in proteomics to simultaneously detect and quantify multiple proteins and their PTMs in complex biological samples. This method leverages the specificity of antibodies to bind to their target antigens, allowing for the high-throughput analysis of protein expression, modifications, and interactions. Below, we elaborate on the principle, applications, and advancements in this technology.
Principle and Mechanism
Antibody microarrays consist of a collection of antibodies immobilized on a solid surface, each specific to a particular protein or PTM. When a sample containing target proteins is applied to the array, these proteins bind to their corresponding antibodies. The binding events are then detected using labeled secondary antibodies or other detection systems, enabling the quantification of bound proteins. This platform can analyze hundreds to thousands of proteins simultaneously, making it an invaluable tool for systems biology studies.
High-Throughput Analysis of Protein Expression
One of the primary applications of antibody microarrays is the high-throughput profiling of protein expression levels. For instance, Haab et al. (2001) demonstrated the utility of this technology in analyzing the expression of various cytokines in cancer patients. The study highlighted the ability of antibody microarrays to identify protein expression patterns associated with specific disease states, providing insights into disease mechanisms and potential biomarkers.
Detection of Post-Translational Modifications
Antibody microarrays are particularly advantageous for studying PTMs, such as phosphorylation, acetylation, and ubiquitination. These modifications play crucial roles in regulating protein function and cellular processes. Anderson and Hunter (2006) utilized antibody microarrays to investigate phosphorylation events in signaling pathways, providing a comprehensive overview of kinase activities in different cell types. The study underscored the sensitivity and specificity of antibody microarrays in detecting low-abundance phosphorylated proteins.
Advantages and Limitations
Advantages:
High Sensitivity and Specificity: The use of specific antibodies ensures high sensitivity and specificity in detecting target proteins and PTMs.
Parallel Analysis: Allows simultaneous analysis of multiple proteins and modifications, providing a holistic view of cellular proteomes.
Quantitative Capability: Offers quantitative data on protein expression levels and modifications, which is critical for understanding biological processes and disease mechanisms.
Limitations:
Antibody Availability: The technology's success depends on the availability and quality of antibodies. The lack of specific antibodies can limit the scope of analysis.
Cross-Reactivity: Antibodies may exhibit cross-reactivity, leading to false positives and inaccuracies in data interpretation.
Dynamic Range: The dynamic range of detection may be limited compared to other proteomic methods, such as mass spectrometry.
Recent Advances and Future Directions
Recent advancements in antibody microarray technology include the development of higher-density arrays, improved detection methods, and enhanced data analysis tools. For instance, Song et al. (2011) reported the integration of fluorescence-based detection with antibody microarrays, significantly improving sensitivity and allowing for the detection of low-abundance proteins. Furthermore, the use of advanced bioinformatics tools has facilitated the analysis of complex data sets, enabling more accurate interpretation of protein expression and modification patterns.
Creative Proteomics continues to advance the field of protein modification analysis by integrating cutting-edge technologies and methodologies. From mass spectrometry-based identification to antibody microarray screening, these techniques offer a comprehensive toolkit for studying the complex landscape of protein modifications. By leveraging these advanced methods, researchers can gain valuable insights into the regulation of protein function and the underlying mechanisms of diseases, paving the way for the development of targeted therapies and diagnostic tools.
References
- Mertins, P., et al. (2016). Proteogenomics connects somatic mutations to signaling in breast cancer. Nature, 534(7605), 55-62.
- Kim, M. S., et al. (2019). A draft map of the human proteome. Nature, 509(7502), 575-581.
- Hornbeck, P. V., et al. (2015). PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Research, 43(Database issue), D512-D520.
- Song, E. Q., et al. (2011). Antibody microarray-based immunoassay for detecting serum protein biomarkers in lung cancer. Journal of Immunological Methods, 364(1-2), 42-49.
- Haab, B. B., et al. (2001). Protein microarrays for highly parallel detection and quantitation of specific proteins and antibodies in complex solutions. Genome Biology, 2(2), research0004.1.
- Anderson, N. L., & Hunter, C. L. (2006). Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Molecular & Cellular Proteomics, 5(4), 573-588.
- Olsen, J. V., Blagoev, B., Gnad, F., Macek, B., Kumar, C., Mortensen, P., & Mann, M. (2006). Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell, 127(3), 635-648.
- Mann, M., Ong, S. E., Grønborg, M., Steen, H., Jensen, O. N., & Pandey, A. (2002). Analysis of protein phosphorylation using mass spectrometry: deciphering the phosphoproteome. Trends in Biotechnology, 20(6), 261-268.
- Cox, J., & Mann, M. (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature Biotechnology, 26(12), 1367-1372.

Related Articals
Our products and services are for research use only.
