
What is Protein Methylation
Protein methylation is a pivotal post-translational modification, wherein methyl groups are covalently attached to specific amino acid residues of proteins, catalyzed by methyltransferases. This process predominantly targets histone and non-histone proteins, focusing mainly on residues of arginine (Arg) and lysine (Lys).
As illustrated in Figure 1, arginine methylation is initiated by protein arginine methyltransferases (PRMTs). Additionally, demethylation of arginine is catalyzed by members of the Jumonji C-terminal (JmjC) domain-containing family, including KDM3A, KDM4E, and KDM5C. Proteins containing Tudor domains and Plant Homeodomain (PHD) zinc fingers recognize arginine methylation and facilitate varied cellular processes dependent on this modification.
Figure 1b elucidates that lysine methylation is mediated by lysine methyltransferases (KMTs). The demethylation of lysine residues is primarily executed by JmjC family members and lysine-specific demethylase 1 (LSD1). The recognition of lysine methylation is conducted by the "Royal" superfamily of domains, comprising Tudor, chromo, malignant brain tumor (MBT), and proline-tryptophan-tryptophan-proline (PWWP) domains, along with PHD zinc fingers. These modules play critical roles in mediating methylation-dependent interactions and functions within cellular contexts.
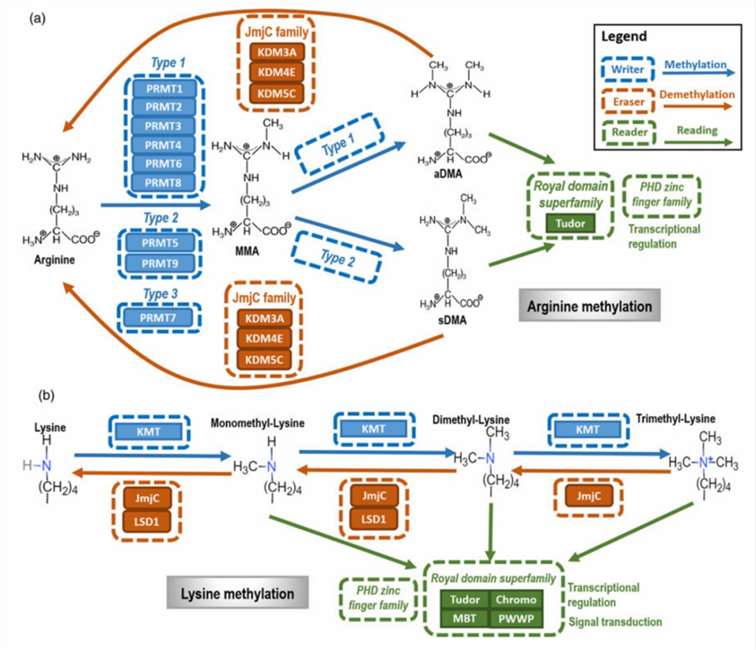 Figure 1. The primary types and mechanisms of protein methylation. (Xiaofeng Dai et al,.2021)
Figure 1. The primary types and mechanisms of protein methylation. (Xiaofeng Dai et al,.2021)
What are Major Types and Mechanisms of Protein Methylation
The following table summarizes the sites, types, involved methyltransferases, and functional roles of Lysine (Lys) and Arginine (Arg) methylation modifications.
For more information, refer to "Mechanism and Research Methods of Arginine Methylation."
Table 1: Histone Methylation Modifications
| Modification Type | Methylation Sites | Methyltransferases | Functions |
|---|---|---|---|
| Lysine Methylation (Kme) | H3: K4, K9, K27, K36; H4: K20 | HKMTs | Stem cell maintenance, differentiation, X chromosome inactivation, transcriptional regulation, DNA damage response |
| Arginine Methylation (Rme) | H3: R2, R3, R7, R26 | PRMT | DNA repair, signal transduction, cell development, cancerogenesis |
| Modification Types | Mono-methylation (me1), Di-methylation (me2), Tri-methylation (me3) | - | Mono-methylation, symmetric and asymmetric di-methylation |
Protein methylation is a reversible modification predominantly occurring in the cell nucleus and on nuclear proteins, often mentioned alongside acetylation as common epigenetic modifications. As a classic epigenetic alteration, histone methylation regulates chromatin structure and gene transcription activity significantly. The methylation of histones is controlled by histone methyltransferases (HMTs) and histone demethylases (HDMs). At least five arginine residues (H3R2, H3R8, H3R17, H3R26, and H4R3) and six lysine residues (H3K4, H3K9, H3K27, H3K36, H3K79, and H4K20) on histones H3 and H4 are known methylation sites. Methylation at different sites may exert both activating and repressive effects on transcription.
Apart from histones, numerous other proteins are subject to methylation. Protein methylation can influence protein-protein interactions, protein-DNA or protein-RNA interactions, protein stability, subcellular localization, or enzyme activity. Methylation modifications of many transcription factors can impact gene expression. Unlike acetylation and phosphorylation, methylation does not alter the total charge of the residue but still affects its properties.
Select Service
Learn more
What are Research Methods of Protein Methylation
The study of protein methylation employs several methodologies to determine modification types and sites.
Antibodies specific to modified histones and Chromatin Immunoprecipitation (ChIP)
Antibodies targeting all known histone modification sites can be procured commercially. These antibodies are utilized in ChIP experiments, wherein cells or tissue samples are cross-linked with formaldehyde to immobilize histones onto chromatin. Subsequent sonication or nuclease treatment fragments the chromatin, yielding short DNA segments. Immunoprecipitation (IP) is then performed using antibodies specific to the desired histone modification. If the presumed modification sites on target genes or promoters are known, PCR can quantify specific modifications at histone sites. Additionally, this method identifies modification sites within conserved promoter regions and elucidates the dynamics of histone modifications at specific sites.
ChIP-on-chip
The ChIP-on-chip technique is utilized to identify genomic regions with specific histone modifications when the modification sites are unknown. In this experiment, DNA fragments IP are labeled with different fluorophores, along with the IP itself. Subsequently, samples are hybridized onto chips containing DNA probes, where the enrichment of a specific DNA fragment in the IP correlates with histone-specific modifications in particular genomic regions.
Mass spectrometry
Mass spectrometry is employed to detect protein methylation and determine the amino acid residues involved in such modifications. The fundamental principle involves using liquid chromatography tandem mass spectrometry to analyze enriched methylated peptide segments. Subsequently, biological information is integrated with mass spectrometry data to identify methylated amino acids, thereby enabling high-throughput detection of protein methylation. The proteomic workflow for methylation modification encompasses protein extraction, enzymatic digestion, enrichment of methylated peptide segments, peptide separation, mass spectrometry analysis, raw data interpretation, and bioinformatics analysis.
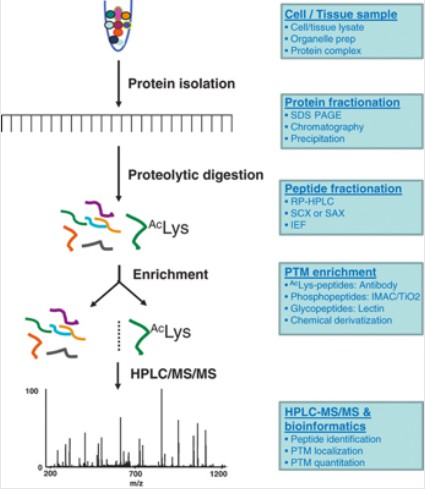 Figure 2. Proteomics Experimental Workflow (Zhao, Y. and Jensen, O. N. 2009)
Figure 2. Proteomics Experimental Workflow (Zhao, Y. and Jensen, O. N. 2009)
Research Strategies for Protein Methylation
In the study of histone methylation modifications, several research avenues are recommended:
Comprehensive Integration of Histone Methylation with Metabolomics: This involves interdisciplinary research linking histone methylation with glycolysis or nucleotide metabolism to comprehensively understand its role in metabolic processes.
Correlative Studies of Existing Histone Modifications and Mechanisms: This includes exploring the association between histone methylation and the mechanisms of diseases such as cancer and inflammation, as well as the combined effects of histone methylation with drug therapies, revealing its potential roles in disease occurrence and treatment.
In-depth Exploration of Histone Biological Functions: For instance, investigating the role of histone methylation in transcriptional regulation to further understand its function in gene expression control.
Specialized Studies on Effector Proteins: This research aims to discover novel modified effector proteins to expand our understanding of histone methylation effector proteins.
In-depth Studies on Related Modifying and Demodifying Enzymes: This includes purification and identification of modifying enzymes to further elucidate the molecular mechanisms of histone methylation modifications.
Case Analysis of Methylation Modifications
The following case analysis explores the methodologies and implications of histone methylation research:
Histone H3 Proline 16 Hydroxylation Regulates Mammalian Gene Expression
Research Techniques:
GST pull-down, immunoprecipitation, His-tagged protein purification, isothermal titration calorimetry (ITC) analysis, mass spectrometry, nuclear magnetic resonance (NMR) spectroscopy.
Research Background:
PTMs of histones play a critical role in regulating various DNA modification processes. In mammalian cells, a specific histone PTM—hydroxylation of proline at position 16 on histone H3 (H3P16oh)—is catalyzed by the prolyl hydroxylase EGLN2. The study identified that H3P16oh enhances the direct binding of KDM5A to H3K4me3, resulting in a concomitant decrease in H3K4me3 at target genes. Further data indicated that, in triple-negative breast cancer (TNBC), the EGLN2-H3P16oh-KDM5A pathway inhibits the WNT pathway negative regulator DKK1, thereby promoting WNT/β-catenin signaling. This study characterized a regulatory mark in the histone code, elucidating the role of H3P16oh in mammalian gene expression regulation.
Previous studies have reported that isomerization of proline at position 38 on histone H3 regulates H3K36 methylation, highlighting the potential significance of histone proline residues in gene regulation. Histone H3 contains six proline residues. Pull-down assays using recombinant FLAG-EGLN1/2/3 proteins and total MDA-MB-231 cell lysates demonstrated that EGLN2 can directly bind to H3. Experimental data revealed that among the first 20 amino acids of H3, only one proline residue—proline 16 (P16)—is present. Consequently, the authors hypothesized that EGLN2 binds to histone H3 through proline 16.
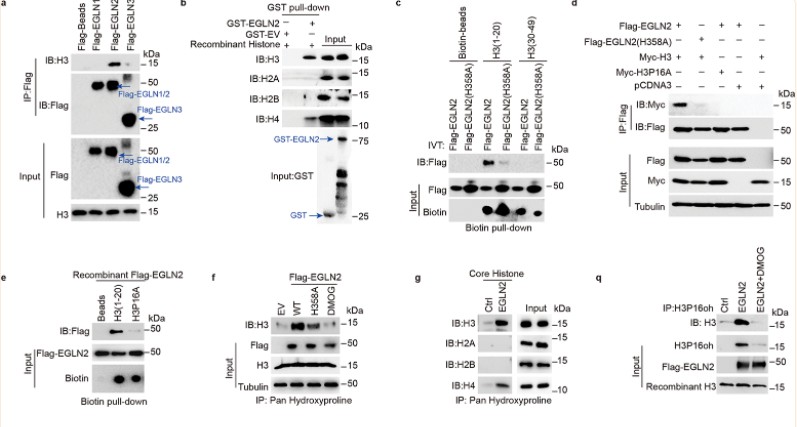 Figure 3. EGLN2 hydroxylates histone H3 on proline 16.
Figure 3. EGLN2 hydroxylates histone H3 on proline 16.
The authors further conducted in vitro hydroxylation assays using various methodologies, demonstrating that EGLN2 facilitates the hydroxylation of proline residues on histone H3.
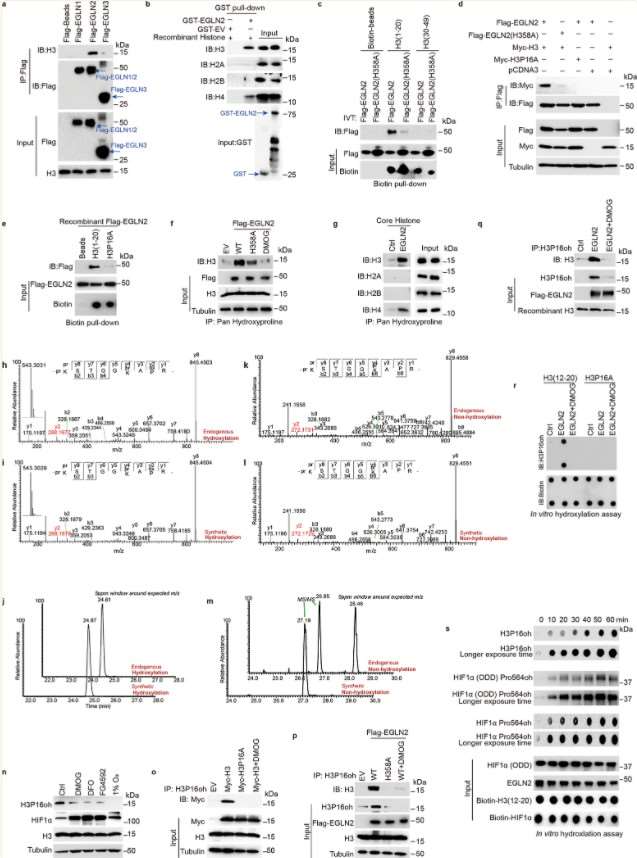 Figure 4. EGLN2 hydroxylates histone H3 on proline 16.
Figure 4. EGLN2 hydroxylates histone H3 on proline 16.
The authors transfected H3 peptide mutants lacking proline (residues 1-20) into cells depleted of EGLN2. They observed that the effect of EGLN2 on H3K4me3 is mediated through the hydroxylation of proline 16 on histone H3 (H3P16oh).
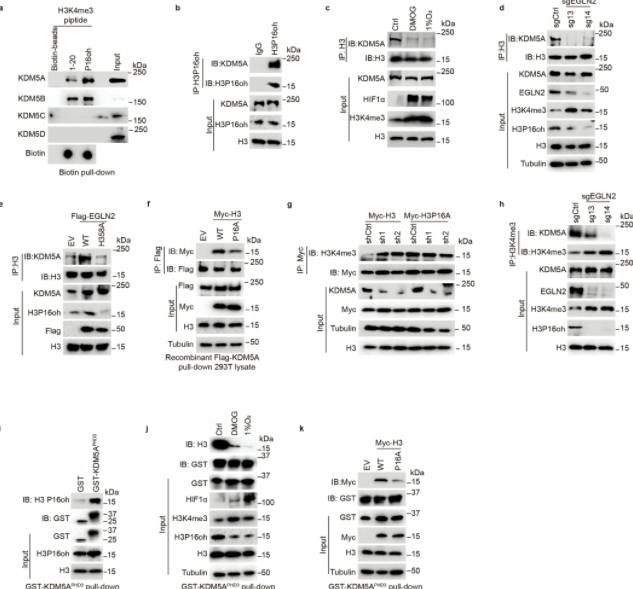 Figure 5. H3P16oh recruits KDM5A and leads to decreased H3K4me3.
Figure 5. H3P16oh recruits KDM5A and leads to decreased H3K4me3.
Subsequent experiments utilizing co-immunoprecipitation (Co-IP), GST pull-down, isothermal titration calorimetry (ITC), and nuclear magnetic resonance (NMR) revealed that the hydroxylation of proline 16 on histone H3 (H3P16oh) enhances the affinity of H3K4me3 for KDM5A, a specific demethylase for H3K4me3. This interaction consequently inhibits the methylation at H3K4.
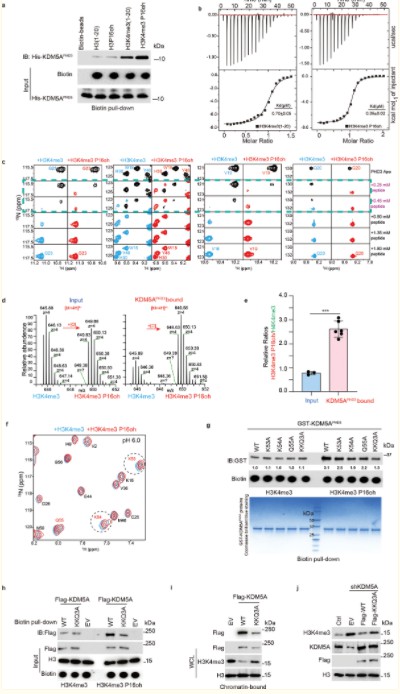 Figure 6. P16oh modification on histone H3 enhances its affinity to KDM5A
Figure 6. P16oh modification on histone H3 enhances its affinity to KDM5A
Subsequently, the authors employed RNA sequencing (RNA-seq) and cleavage under targets and release using nuclease (CUT&RUN) techniques to investigate the genome-wide functions of H3P16oh. The integrated genomic and transcriptomic analyses identified a series of direct targets of the EGLN2-H3P16oh-KDM5A axis.
Further exploration revealed that EGLN2-mediated H3P16oh modification inhibits the distribution of H3K4me3 at the promoter region of DKK1, thereby repressing DKK1 transcriptional activation. DKK1 is a negative regulator of the WNT signaling pathway, and its transcriptional repression leads to the activation of WNT signaling, a critical factor in the pathogenesis of breast cancer. The expression levels of EGLN2, H3P16oh, and H3K4me3 were examined across various murine tissues, revealing a correlation in their expression patterns. EGLN2-catalyzed H3P16oh enhances the recruitment of KDM5A and influences genome-wide H3K4me3 distribution in breast cancer cells, thereby modulating the KDM5A-H3K4me3-DKK1-WNT signaling axis in these cells.
The authors also subjected triple-negative breast cancer (TNBC) cells to hypoxic conditions (1% O2) and performed CUT&RUN assays. Under normoxic conditions (20% O2), H3P16oh and H3K4me3 were co-distributed across 8,294 genes in the genome. Under hypoxic conditions, the distribution of H3P16oh at these 8,294 genes decreased, while H3K4me3 levels increased. These data indicate that H3P16oh negatively regulates H3K4me3 under pathological conditions.
Finally, genome-wide and transcriptomic analyses demonstrated that the knockout of EGLN2 inhibits the proliferation of TNBC cells, whereas it does not significantly affect the growth of 293T cells. The authors hypothesize that this discrepancy may be due to the differential regulation of genes by the EGLN2-H3P16oh axis in distinct pathological and cellular contexts.
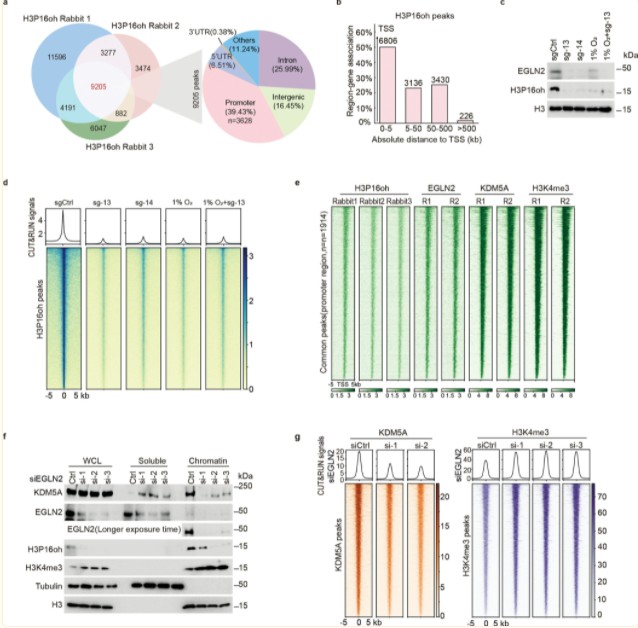 Figure 7. EGLN2-catalyzed H3P16oh enhances recruitment of KDM5A and affects H3K4me3 genome-wide in breast cancer cells.
Figure 7. EGLN2-catalyzed H3P16oh enhances recruitment of KDM5A and affects H3K4me3 genome-wide in breast cancer cells.
The research outlined in this study elucidates the role of EGLN2 in hydroxylating H3P16me3 to produce H3P16oh, thereby regulating the KDM5A-H3K4me3-DKK1-WNT signaling axis in breast cancer cells. Additionally, it demonstrates that oxygen sensing signaling pathways can directly influence H3P16oh modification.
References
- Susan C. Wu, Yi Zhang, Minireview: Role of Protein Methylation and Demethylation in Nuclear Hormone Signaling, Molecular Endocrinology, Volume 23, Issue 9, 1 September 2009, Pages 1323–1334.
- Zhang, D., Guo, W., Wang, T., Wang, Y., Le, L., Xu, F., Wu, Y., Wuriyanghan, H., Sung, Z. R., Pu, L., RNA 5-Methylcytosine Modification Regulates Vegetative Development Associated with H3K27 Trimethylation in Arabidopsis. Adv. Sci. 2022, 10, 2204885.

Related Articals
Our products and services are for research use only.
