The Label-Free quantitative proteomics technology, which offers simple experimental procedures, unlimited sample numbers and types, and strong data reproducibility, demonstrates excellent capabilities in quantifying changes in protein abundance and off-target effects induced by compounds.
Label-Free quantification is a method for determining the relative abundance of proteins in two or more biological samples. Unlike other quantification methods, Label-Free does not use stable isotopes for chemical binding and labeling of proteins. Label-Free quantitative proteomics provides a powerful tool for resolving and identifying thousands of proteins from complex biological samples. In this method, proteins are first digested into peptide mixtures using proteases and then detected and analyzed through tandem mass spectrometry (MS/MS) and database searching. Compared to other proteomic methods, Label-Free quantification is faster and more sensitive, increasing the dynamic range of protein detection by 3 to 4 times compared to 2DE. This method can also be automated, enabling the analysis of large batches of samples for differential protein level analysis.
Label-Free Quantitative Proteomics Technical workflow
Label-Free Quantitative Proteomics Service Advantages
(1) The experimental procedure is relatively simple.
(2) There is no limit to the number of samples.
(3) It is suitable for various types of samples.
(4) The quality control process is comprehensive.
(5) The data reproducibility is excellent.
Label-Free Quantitative Proteomics Applications
(1) Quantify protein abundance and identify disease biomarkers or drug targets.
(2) Deepen understanding of the mechanism of action (MOA) of drugs.
(3) Uncover drug therapeutic effects (such as the degradation of target proteins by PROTAC) and potential adverse effects (such as off-target effects of PROTAC drugs).
Label-Free Quantitative Proteomics Applications in PROTAC Drug Development
(1) Changes in protein abundance and off-target effects caused by the compound.
(2) Utilize the advantages of mass spectrometry-based absolute quantification to determine the activity of the compound in complex biological environments (such as whole blood PBMC, tissues, cells, etc.) in degrading target proteins (calculate EC50/90, Dmax, etc.).
(3) Determine the degradation level of affinity proteins for the target protein to facilitate the design and evolution of highly selective small molecules.
(4) Gain insights into the degradation synthesis rate (mPDP) of the target protein through mass spectrometry analysis.
Classic case
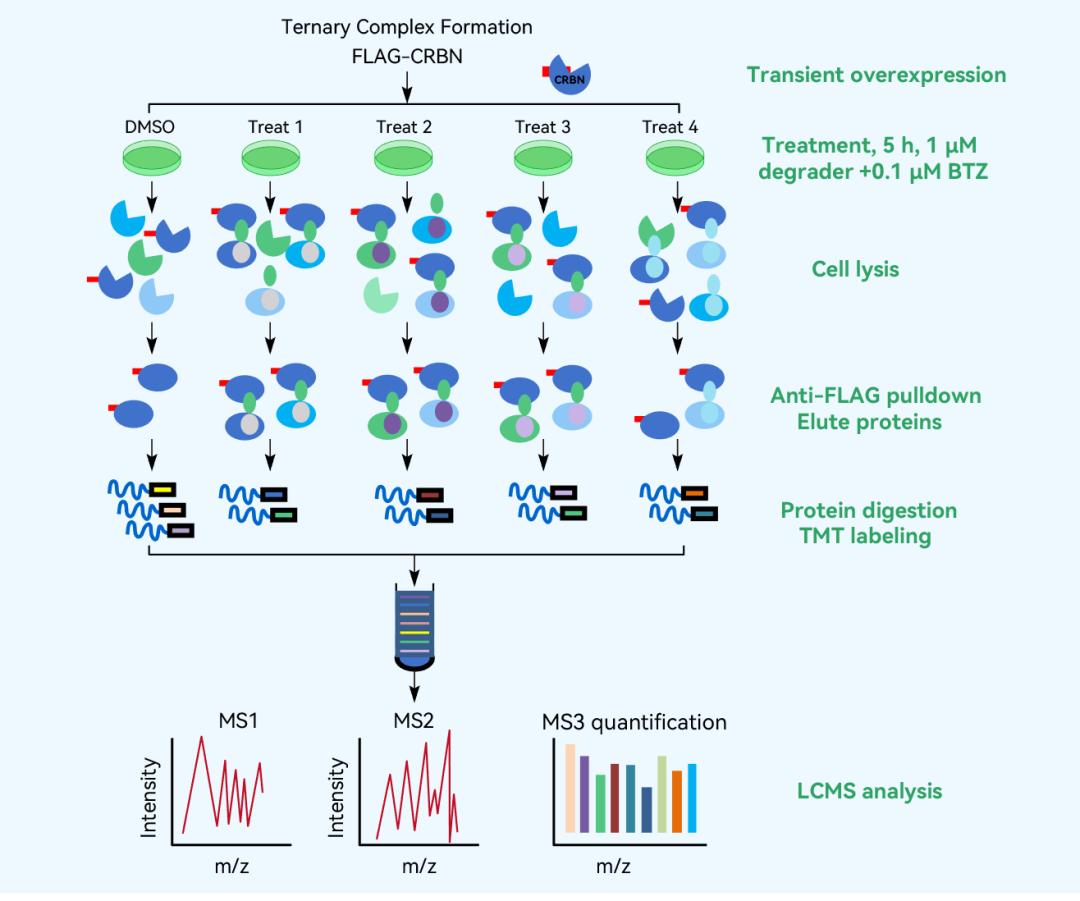
Relative abundance ratios of enriched proteins in the FLAG-CRBN AP-MS experiment were validated and sorted.
A. Workflow schematic of quantitative cell proteomics. B. Proportions of quantified proteins and contributions from different cell lines.
Our PROTACs Degradation Omics Solution
Creative Proteomics utilizes the following techniques to analyze PROTAC degradation:
(1) Extensive non-targeted protein detection using DIA and TMT.
(2) Targeted quantitative detection using MRM/PRM.
(3) AP-MS for protein-protein interaction analysis.
(4) Ubiquitin proteomics for studying ubiquitination-mediated protein degradation.
(5) Protein quantification using Simple/Digital Western technology.
Reference
- Liu X, Zhang Y, Ward LD, Yan Q, Bohnuud T, Hernandez R, Lao S, Yuan J, Fan F. A proteomic platform to identify off-target proteins associated with therapeutic modalities that induce protein degradation or gene silencing. Sci Rep. 2021 Aug 4;11 (1):15856.
Services You May Be Interested In
Our products and services are for research use only.